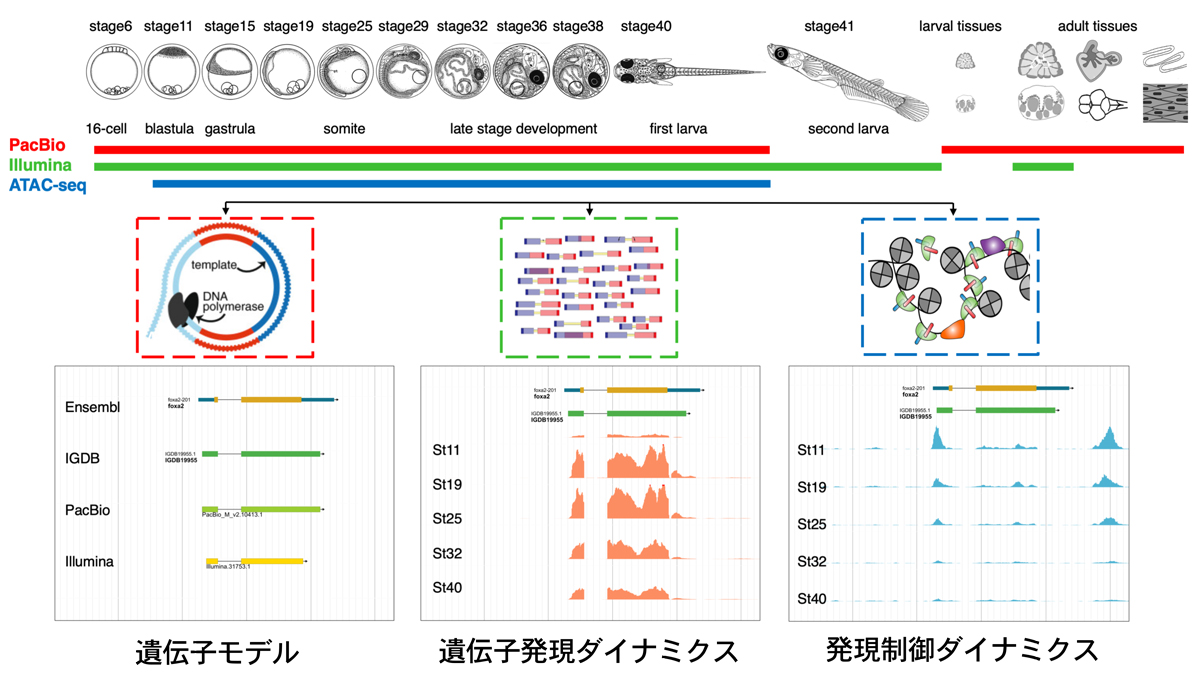
メダカは遺伝学、発生学、環境科学から基礎医学まで様々な研究分野で世界的に利用されています。系統的に大きく異なる3近交系の高精度ゲノム塩基配列も決定され、遺伝子導入・ゲノム編集などを含む様々な遺伝的手法も開発されています。しかし、現在公表されているメダカゲノムの注釈(Genome Annotation)は、計算機による遺伝子予測とshort-read RNA-seqに基づく遺伝子構造推定を組み合わせた遺伝子モデルが中心であり、Genome Annotationの間違いもかなり含まれています。また発生に伴うと遺伝子発現量とChromatin Accessibilityの関係も発生段階全体を通じた解析はありませんでした。より正確な遺伝子モデルとその発現変化及びChromatin Accessibilityの関係を詳細に記載することはメダカの利用をさらに促進するための重要なステップとなります。中国科学院遺伝・発育生物学研究所のQiang Tu博士らと基礎生物学研究所の成瀬清特任教授の国際共同研究チームは、11の異なる発生段階の胚と稚魚・成魚の各臓器を含む19種類のサンプルより新たにRNAを抽出し長鎖RNA-seq法(long-read RNA-seq)によって完全長cDNA配列に基づく新しい遺伝子モデルを構築しました。さらに発生段階ごとにshort-read RNA-seqとtransposase-accessible chromatin using sequencing (ATAC-Seq) を行い、各遺伝子の発生に伴う遺伝子発現量変化とChromatin Accessibilityの変化を調べることで、同定した全遺伝子にわたる新たなGenome Annotationを行いました。研究チームは、これら一連のGenome Annotation解析系をMinimum ENCODE toolboxと名付けました。Minimum ENCODE toolboxによる解析の結果、17,000 の新規isoforms、1,600の転写因子、 1,100 のlong non-coding RNAsを含む26,548の遺伝子及び150,000のシス調節領域を同定しました。またこれらのデータを小型魚類コミュニティーに公開するため、新たにMedaka omics data portal (
http://tulab.genetics.ac.cn/medaka_omics)を構築しました。このサイトでは、Genome Browser (JBrowse)とともにGene Viewer (Shiny)(発生に伴う遺伝子発現量変化)及びBLASTによる配列相同性による遺伝子検索の3つのツールを備えています。さらに今回の研究で明らかになったメダカゲノム注釈データに加えて、過去に発表された他のOmicsデータ(RNA-seq, ATAC-seq, ChIP-seq)についても同時にゲノムブラウザーで参照することが可能です。この成果はGenome Research誌に掲載されました。
本研究の概要図