メダカは遺伝学、発生学、環境科学から基礎医学まで様々な研究分野で世界的に利用されています。系統的に大きく異なる3近交系の高精度ゲノム塩基配列も決定され、遺伝子導入・ゲノム編集などを含む様々な遺伝的手法も開発されています。しかし、現在公表されているメダカゲノムの注釈(Genome Annotation)は、計算機による遺伝子予測とshort-read RNA-seqに基づく遺伝子構造推定を組み合わせた遺伝子モデルが中心であり、Genome Annotationの間違いもかなり含まれています。また発生に伴うと遺伝子発現量とChromatin Accessibilityの関係も発生段階全体を通じた解析はありませんでした。より正確な遺伝子モデルとその発現変化及びChromatin Accessibilityの関係を詳細に記載することはメダカの利用をさらに促進するための重要なステップとなります。中国科学院遺伝・発育生物学研究所のQiang Tu博士らと基礎生物学研究所の成瀬清特任教授の国際共同研究チームは、11の異なる発生段階の胚と稚魚・成魚の各臓器を含む19種類のサンプルより新たにRNAを抽出し長鎖RNA-seq法(long-read RNA-seq)によって完全長cDNA配列に基づく新しい遺伝子モデルを構築しました。さらに発生段階ごとにshort-read RNA-seqとtransposase-accessible chromatin using sequencing (ATAC-Seq) を行い、各遺伝子の発生に伴う遺伝子発現量変化とChromatin Accessibilityの変化を調べることで、同定した全遺伝子にわたる新たなGenome Annotationを行いました。研究チームは、これら一連のGenome Annotation解析系をMinimum ENCODE toolboxと名付けました。Minimum ENCODE toolboxによる解析の結果、17,000 の新規isoforms、1,600の転写因子、 1,100 のlong non-coding RNAsを含む26,548の遺伝子及び150,000のシス調節領域を同定しました。またこれらのデータを小型魚類コミュニティーに公開するため、新たにMedaka omics data portal (
http://tulab.genetics.ac.cn/medaka_omics)を構築しました。このサイトでは、Genome Browser (JBrowse)とともにGene Viewer (Shiny)(発生に伴う遺伝子発現量変化)及びBLASTによる配列相同性による遺伝子検索の3つのツールを備えています。さらに今回の研究で明らかになったメダカゲノム注釈データに加えて、過去に発表された他のOmicsデータ(RNA-seq, ATAC-seq, ChIP-seq)についても同時にゲノムブラウザーで参照することが可能です。この成果はGenome Research誌に掲載されました。
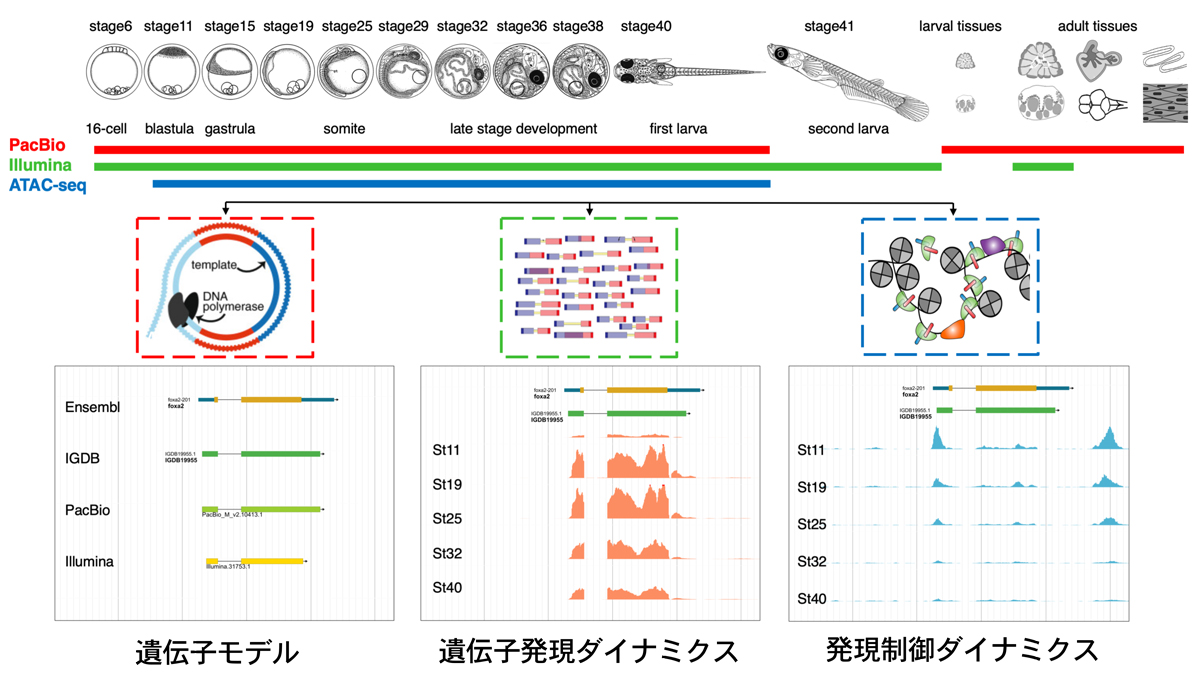
本研究の概要図
【研究の背景】
メダカは遺伝学、発生学、進化学、環境科学から基礎医学分野まで様々な研究に用いられています。2007年にゲノム概要配列が公開され、2016年には3系統の近交系を用いてPacific Bioscience 社のsingle-molecule real-time (SMRT) テクノロジーを用いた高精度のゲノム塩基配列が公開されています。ゲノム配列の解析ではゲノム塩基配列とともにその注釈付け(Genome Annotation)が重要です。ヒトやマウスゲノムではGenome Annotationのための国際プロジェクトであるENCODEプロジェクトが実施されその結果が公開され多くの研究者に利用されています。一方で、現在公開されているメダカゲノムのGenome Annotationは計算機を用いた予測遺伝子と比較ゲノム解析データに加え、EST解析やshort-read RNA-seqによるデータを加味した遺伝子モデルが構築され、それに基づいたGenome Annotationがおこなわれています。その結果、転写配列へのイントロン配列の混入や同じ遺伝子座に由来する二種類のisoformが融合した形で予測されるなどGenome Annotationの間違いもかなり見受けられる状態でした。また核遺伝子の発現量と近傍のChromatin Accessibilityの関係を発生段階全体にわたって解析したデータもありませんでした。従来のGenome Annotationの欠点を補い、生物学的な事実に基づくより正確なGenome Annotationを行うことはメダカの利用をさらに促進するためには不可欠です。そのため、研究チームはSMRTテクノロジーに基づくlong-read RNA-seqを用いて完全長cDNAに基づく新たな遺伝子モデルを作成し、Ensembl遺伝子モデルと比較することで新しい遺伝子モデル(IGDB)を構築しました。さらにメダカの発生に伴う遺伝子発現変化を明らかにするためIllumina short-read RNA-seqによる遺伝子発現量の解析と発生に伴うクロマチン状態の変化を明らかにするためATAC-seqによるChromatin Accessibility解析を行いました。
【研究の成果】
研究チームは、11の異なる発生段階の胚と稚魚・成魚の各臓器からなる19種類のサンプルより新たに高品質なRNAを抽出し、cDNAライブラリーを作成しました。そして、Pacific Bioscience 社のsingle-molecule real-time (SMRT) テクノロジーを用いたlong-read RNA-seq法により完全長cDNAの塩基配列を決定しました。その結果、28,427の転写産物よりなる17,523遺伝子を同定しました。この遺伝子セットをEnsemblで予測されている38,187の転写産物よりなる24,366の遺伝子セットとの比較を行いshort-read RNA-seqのデータを用いた遺伝子発現情報を加味することで新たに40,960の転写産物よりなる26,548遺伝子からなる新規メダカ遺伝子モデル(IGDB)を構築しました。
研究チームは、この新規遺伝子モデルの中でも転写因子とlong non-coding RNAs (lncRNAs)に注目しました。転写因子は他の遺伝子の発現調節を担う遺伝子であり、生命過程の‘master regulators’ 或いは ‘selector genes’としての働きを持っています。今回の解析で68遺伝子ファミリーからなる1,646の転写因子が同定されました。lncRNAsは200bpを越える長さを持ち5’末にCap構造を、そして3‘端にポリA構造をもつ典型的なmRNA構造を持ちます。しかしlncRNAsはタンパク質コード領域を含まず、タンパク質への翻訳が起こらないRNAとして近年注目されています。今までのlncRNAsの同定は、short-read RNA-seqを用いた解析が多く、その構造解析ではアセンブリープロセスによる構造推定が含まれることから、配列にエラーを含む場合がありました。今回用いたlong-read RNA-seq法では長いcDNAであっても連続した一連の塩基配列として全構造を決定できることから、アセンブリープロセスを経ることなくcDNAの塩基配列を決定できるという長所を持っています。そのため、解析から同定されたlncRNAs配列は、より配列エラーが少ない新たなlncRNAsデータとして利用できます。この解析から1,135のlncRNAsが同定されました。一般にlncRNAsは種特異性が高く種間での保存性が低いことが知られています。今回の解析でも、8つのlncRNAsのみがヒトlncRNAsと相同性(e-value <10
-10)を示しました。またゼブラフィッシュとの間でもわずか10のlncRNAsのみが相同性(e-value <10
-10)を示しました。
次にこれらの遺伝子の、発生にともなう遺伝子発現量の変化と当該遺伝子周辺のクロマチン状体との関係を調べました。9の異なった発生段階の胚よりRNAを抽出し、short-read RNA-seqにより構築した全遺伝子の、発生に伴う遺伝子発現量変化を明らかにすると同時に、同じ発生段階の胚のDNAを用いてATAC-seqにより発生に伴うクロマチン状態の変化について同時に解析しました。ATAC-seq法はクロマチン状態が開いている(転写因子等の発現調節因子が結合しやすい状態であると考えられている)ゲノム領域を特異的に同定する方法です。この二つの方法を併用することで、特定の発生段階で発現している遺伝子の発現量と当該遺伝子周辺のクロマチン状態との関係を調べることができます。
その結果、今回作成した26,548遺伝子よりなるIGDBのうち79%はメダカの発生過程において発現(1TPMレベル以上)していました。各遺伝子の発現プロファイル(発生に伴う遺伝子発現量の変化)について分類するためMfuzz法を用いてクラスタリングしたところ30の異なるクラスターに分けることができました。特定のクラスターは特徴的なGO termに分類される遺伝子がenrichしていることも明らかとなりました。これは転写の開始、中胚葉誘導、神経発生など発生段階の変遷に伴い発現する遺伝子の変化に由来すると考えられます。
従来の研究から転写開始点近傍は遺伝子発現に重要なシス制御領域が多く存在することが知られています。そこでATAC-seqにより得られたゲノム配列のタグの位置を解析したところ、明らかに転写開始点付近にenrichしていました。この結果から、今回作成されたATAC-seqライブラリーはシス制御領域を上手く補足していると考えられます。
遺伝子の発現量とそのシス制御領域のChromatin Accessibilityとの関係を明らかにするためにはどの領域が当該遺伝子の制御に関与するのかを明らかにする必要があります。多くの発現制御領域は最近傍の遺伝子の制御をおこなうことが知られています。そこでATAC-seqによって得られた発現制御領域のChromatin Accessibility(ATAC-seqによって得られたTag数)と最近傍の遺伝子の発現量の変化(RNA-seqで得られたTag数)を調べました。
その結果、遺伝子発現量と制御領域のChromatin Accessibilityとの関係はおおよそ4つのパターンが分けられることが明らかとなりました。第1のタイプはsynchronization(相引)です。このタイプでは発現量の増減と近傍のシス制御領域のChromatin Accessibilityの増減が高い正の相関を示します。第2のタイプはrepression(相反)です。このタイプでは制御領域のChromatin Accessibilityが高いと遺伝子の発現量が顕著に抑制されます。このような制御を受ける遺伝子ではその制御領域はインシュレータ配列に多くみられるCTCF と CTCFLモチーフがenrichしていました。第3のタイプはenhancer switching(制御領域スイッチ)です。このタイプの遺伝子では発生の初期では領域AのChromatin Accessibilityと遺伝子発現量が正の相関を持ち、後期では別の領域BのChromatin Accessibilityとその発現量が正の相関を持っていました。このような遺伝子では発生に伴って制御領域がスイッチしていること考えられる。第4のパターンはearly opening(前開始)です。このパターンでは制御領域のChromatin Accessibilityの増加にかなり遅れて遺伝子発現量が増加します。発生では3,000を越える遺伝子がこのようなパターンを持つことが明らかとなりました。
研究チームは、PacBio long-read RNA-seq法による遺伝子モデルの構築、Illumina short-read RNA-seq法による遺伝子発現推定、ATAC-seq法による発現制御領域のChromatin Accessibility解析をくみ交わせたmulti-omics解析プラットフォームをMinimum ENCODE toolboxと名付けました。一連の解析によってはじめてメダカのほぼすべての発生段階での全遺伝子レベルの遺伝子発現プロファイルと近傍の遺伝子制御領域のChromatin Accessibilityを明らかにすることができました。これらのすべてのデータはMedaka omics data portal (
http://tulab.genetics.ac.cn/medaka_omics/)として3つのウエッブツール(Jbrowserを用いたゲノムブラウザー、各遺伝子の機能予測と発現量変化を知ることができる遺伝子ビューワー、配列による相同性検索が可能なblast検索)を用いて小型魚類コミュニティーに公開されています。Medaka omics data portalでは今回の研究で得られたデータに加えて、すでに発表さえている様々なOmicsデータを加えて100以上のデータトラックについても閲覧が可能になっています。
【今後の展望】
中国科学院遺伝・発育生物学研究所のQiang Tu博士は「本研究によってはじめてメダカの発生に伴う遺伝子発現量とクロマチン状態を知るための基礎的データを提供することができました。メダカを用いたオミックス解析では胚単位の解析から単一細胞を用いたmulti omics解析を計画しています。またMinimum ENCODE toolboxを用いてメダカ近縁種にも用いることで進化過程での遺伝子発現制御機構の変化についても基礎的なデータを得ることができます。」と語っています。
基礎生物学研究所の成瀬清特任教授は「今回構築したMinimum ENCODE toolboxは、今後展開されるユニークな生物学的特徴を持つ新規モデル生物を用いた網羅的遺伝子機能解析を行うプラットフォームとしても重要な役割を果たすと考えています」と語っています。
【発表雑誌】
雑誌名 Genome Research
掲載日 2020年6月26日
論文タイトル: Dynamic Transcriptional and Chromatin Accessibility Landscape of Medaka Embryogenesis
著者: Yingshu Li, Yongjie Liu, Hang Yang, Ting Zhang, Kiyoshi Naruse and Qiang Tu
DOI:
https://doi.org/10.1101/gr.258871.119
【研究グループ】
Yingshu Li
1,2,3,+, Yongjie Liu
1,2,3,+, Hang Yang
1,2,3, Ting Zhang
1,2, Kiyoshi Naruse
4, Qiang Tu
1,2,3
1 State Key Laboratory of Molecular Developmental Biology, Institute of Genetics and
Developmental Biology, Innovation Academy for Seed Design, Chinese Academy of Sciences,
Beijing 100101, China.
2 Key Laboratory of Genetic Network Biology, Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Beijing 100101, China
3 University of Chinese Academy of Sciences, Beijing 100049, China.
4 Laboratory of Bioresources, National Institute for Basic Biology, Okazaki 444-8585, Aichi, Japan.
【研究サポート】
本研究はNational Natural Science Foundation of China (31671493, 91740109)、CAS Strategic Priority Research Program (XDA16020801)、自然科学研究機構戦略的国際研究交流加速事業(NINS international Research Collaboration Program 2015)のサポートを受けて実施されました。
【本研究に関するお問い合わせ先】
基礎生物学研究所バイオリソース研究室
特任教授 成瀬 清
TEL: 0564-55-7580/0564-59-5932
E-mail: naruse@nibb.ac.jp
【報道担当】
基礎生物学研究所 広報室
TEL: 0564-55-7628
FAX: 0564-55-7597
E-mail: press@nibb.ac.jp