基礎生物学研究所


2015.03.06
基礎生物学研究所・統合神経生物学研究部門の新谷隆史准教授と野田昌晴教授らは、APC2 (Adenomatous polyposis coli 2)という脳神経系に発現する分子の機能を明らかにする研究を進めています。今回、同グループはカナダのMcGill大学、並びにカタールのHamad Medical Corporationの研究グループとの共同研究により、APC2遺伝子の機能不全が、ソトス症候群と呼ばれる先天性奇形症候群の代表的な症状である知的障害や頭部の過成長を説明することを明らかにしました。この成果は3月6日にCell Reports誌に掲載されました。
[研究の背景]
ソトス症候群(Sotos syndrome)は脳性巨人症とも呼ばれ、知的障害、特徴的な顔貌(大きな頭部や突出した額等を伴う)、幼少期から小児期にかけての過成長、骨年齢の促進、心臓や腎臓の障害などを有する先天性奇形症候群であり、過成長症候群(Overgrowth syndrome)の中では最も頻度が高いものの一つです。新生児の14,000人に1人の割合でソトス症候群の子供が生まれると報告されていますが、未診断の患者も含めると実際には5,000人に1人に近い頻度でソトス症候群の子供が生まれているのではないかと考えられています。
これまでの研究によって、NSD1遺伝子のハプロ不全(2本の染色体上にそれぞれ存在する遺伝子の一方の異常によって、必要とされる機能タンパク質の量が不足すること)がソトス症候群の原因とされています。NSD1遺伝子は、DNAに結合するタンパク質であるヒストンをメチル化修飾する酵素(ヒストンメチル化酵素)をコードしています。ヒストンはDNAを小さく折りたたんで核内に収納する役割を果たしており、NSD1タンパク質は、ヒストンの修飾を通じてDNAの状態を変化させることによって、複数の特定の遺伝子の発現を制御していると考えられています。ソトス症候群においては、NSD1の変異によって、これらの下流の遺伝子の発現に変化が生じ、その結果ソトス症候群に特有の症状が生じると考えられます。しかしながら、NSD1が制御している下流の遺伝子については全く分かっていませんでした。
[研究の成果]
今回、ソトス症候群に特有の症状である知的障害並びに頭部の過成長等を示す海外在住の姉弟について遺伝子解析を行いました。しかしながら、NSD1遺伝子には異常が見出されませんでした。そこで全エクソン解析法(タンパク質をコードする遺伝子領域(エクソン)の配列を全て解読する手法)を用いて調べたところ、APC2遺伝子にホモ(染色体上に存在する二つの遺伝子の両方)のフレームシフト変異(遺伝子塩基配列中の欠失や挿入によって遺伝暗号の読み枠がずれる変異)が、両患者に共通して存在することが明らかになりました(図1)。患者の両親はいとこ同士の関係であることから、両親の遺伝子を調べたところ、両親がヘテロにこの変異を持っていることが判りました。
APC2タンパク質は、細胞の形態や運動、細胞の情報伝達において、重要な役割を果たしている細胞骨格の働きを制御しています(Shintani et al., 2009, 2012)。細胞生物学的解析から、患者の変異APC2タンパク質は、細胞骨格に対する制御能を失った機能欠失型変異体であることが分かりました。そこでApc2遺伝子欠損マウス(APC2の発現を欠失したマウス)が患者と同様な異常を示すかどうかを調べました。まず、行動学的解析を行ったところ、患者と同様に、記憶・学習能に障害があることが明らかになりました。また、患者と同様に、脳室の拡大や頭部骨格の拡大等の異常が見出されました。以上の結果から、患者に見られる知的障害や言語障害、頭部構造の異常は、APC2の機能欠失によって生じたと結論付けられました。
このようにAPC2遺伝子に変異を有する患者がソトス症候群に特有の症状を示すことから、APC2はNSD1の下流遺伝子である可能性が強く示唆されました。そこでこの可能性について検討しました。まず、培養したマウスの神経細胞においてNsd1の発現を人為的に抑制したところ、予想通りApc2の発現が減少しました(図2)。Apc2遺伝子欠損マウスでは、脳の神経細胞の移動に異常があることが判っていました(Shintani et al., 2012)。そこで、発生途中のマウスの大脳皮質の神経細胞においてNsd1の発現を減少させたところ、新生の神経細胞の移動が障害されました(図3)。このような細胞移動の異常はApc2遺伝子欠損マウスだけでなく、Apc2の発現を減少させた場合においても観察されます(図3)。また、Nsd1の発現を減少させた状態で同時にApc2の発現を補ってやると、細胞移動の異常が大きく改善されました(図3)。これらの結果は、APC2がNSD1の下流遺伝子であることを示しています。
APC2は脳神経系に特異的に発現する分子であり、ソトス症候群における神経系に関連した知的障害や頭部の過成長等の症状は、APC2の発現低下が主な原因となっていると考えられます。ソトス症候群では神経系の異常だけでなく、骨年齢の促進、心臓や腎臓の障害などが観察されますが、APC2遺伝子に変異を有する患者においては、これらの異常は見られませんでした。神経系以外のこれらの症状には、APC2以外のNSD1の下流遺伝子が関与していると推測されます(図4)。
APC2は、細胞移動だけでなく、神経細胞の形態形成、神経回路形成や、シナプス可塑性などに関与すると推定されます。今後は、APC2のこれらの機能的役割の中で最も直接的に知的障害に関係しているものを明らかにしていく必要があります。一方、ソトス症候群の良い疾患動物モデルはこれまでにありませんでしたがApc2遺伝子欠損マウスは、ソトス症候群を研究する上で有用なモデル動物になると考えられます。
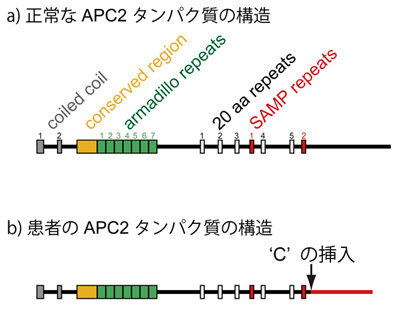
図1 正常なAPC2タンパク質と患者のAPC2タンパク質の構造
患者のAPC2遺伝子にはCの一塩基挿入があるためにそれ以降の遺伝暗号の読み枠がずれ、赤色で示した部分が異常なアミノ酸配列に変わっています。
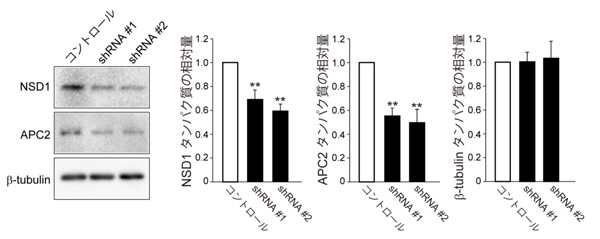
図2 培養したマウスの神経細胞においてNSD1の発現を減少させた場合のAPC2の発現量の変化
培養した神経細胞にNSD1の発現を減少させる遺伝子(shRNA#1と#2)を発現させると、NSD1タンパク量が減少します。それに呼応して、APC2タンパク質の発現量も減少します。この時、NSD1に関係のないb-tubulin量には変化はありません。
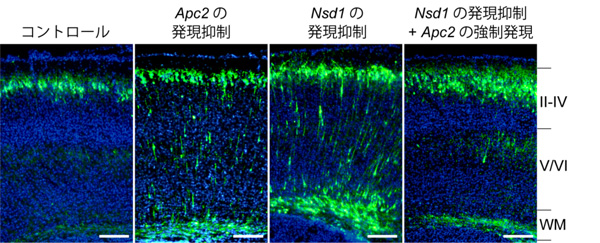
図3 大脳皮質の神経細胞における遺伝子の発現抑制実験
発生15日目にApc2やNsd1の発現を抑制するための遺伝子導入を行った後、生後7日目に解析しました。緑色の細胞が遺伝子導入された細胞。コントロールではほとんどの細胞がII-IV層に移動するのに対して、Apc2やNsd1の発現を抑制した場合にはV/VI層や白質(WM)に留まるような異常が観察されます。一方、Nsd1の発現を抑制した場合にApc2の発現を補うと、異常な移動は改善します(スケールバーは100マイクロメートル)。
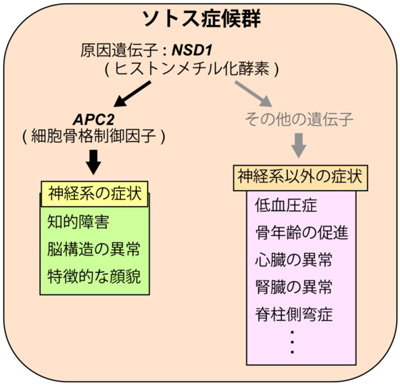
図4 ソトス症候群の発生機序
神経系得的に発現するAPC2遺伝子がNSD1の下流遺伝子であることが明らかになりました。ソトス症候群においては、NSD1遺伝子の異常によってAPC2の発現が減少しており、その結果、知的障害や頭部の過成長等の異常が生じると考えられます。神経系以外の症状には、他のNSD1の下流遺伝子が関与していることが予想されます。
[用語解説]
ソトス症候群:
ソトス症候群は胎生期・小児期に顕著な過成長、特徴的な顔貌、学習障害といった中核となる3つの特徴の他、行動障害、先天性の心奇形、新生児黄疸、腎奇形、脊柱側彎、てんかん発作など多様な症状を来す常染色体優性遺伝性疾患である。別名:脳性巨人症。染色体5q35に位置する「核内受容体結合SETドメイン保有蛋白質遺伝子(NSD1)」が染色体の微細欠失または点変異によってハプロ不全となることでソトス症候群が発症する。NSD1はヒストンの修飾や転写調節に関わると考えられるが機能の詳細は不明である。本邦ではNSD1領域微細欠失の頻度が諸外国と比して高く、NSD1領域のプローブを用いたFISH法による染色体診断が有効と考えられる。現在のところ根治療法はない。
(難病情報センターホームページ(2015年2月現在)から引用)
[掲載紙情報]
Cell Reports(米国東部時間3月5日昼12時にオンライン掲載予定)
論文タイトル:Loss-of-Function Mutation in APC2 Causes Sotos Syndrome Features
著者:Mariam Almuriekhi*, Takafumi Shintani*, Somayyeh Fahiminiya*, Akihiro Fujikawa, Kazuya Kuboyama, Yasushi Takeuchi, Zafar Nawaz, Javad Nadaf, Hussein Kamel, Abu Khadija Kitam, Zaineddin Samiha, Laila Mahmoud, Tawfeg Ben-Omran, Jacek Majewski & Masaharu Noda
*Co-first authors
[研究サポート]
本研究は、文部科学省科学研究費助成事業等の支援を受けて行われました。本研究グループは、基礎生物学研究所ボトムアップ型国際共同研究事業がサポートしています。
[研究グループ]
本研究における各研究グループの研究分担は以下の通りです。
・カタールのHamad Medical Corporation:患者の診断
・カナダのMcGill大学:患者遺伝子の全エクソン解析
・基礎生物学研究所:変異APC2タンパクの機能解析、Apc2遺伝子欠損マウスを用いた解析、
APC2とNSD1の相互関係の解析
・論文作成は基礎生物学研究所のグループが主導しました。
[本研究に関するお問い合わせ先]
基礎生物学研究所 統合神経生物学研究部門
教授:野田 昌晴 (ノダ マサハル)
TEL: 0564-59-5846
E-mail: madon@nibb.ac.jp