基礎生物学研究所


2012.11.08
基礎生物学研究所・統合神経生物学研究部門(野田 昌晴 教授)とアスビオファーマ株式会社の研究グループは、Ptprzというタンパク質チロシン脱リン酸化酵素が、中枢神経系における髄鞘(ミエリン鞘)註1)の形成/再形成の制御に関わることを明らかにしました。研究グループは、Ptprzを欠失させたマウスでは、発生期の脳内において髄鞘の形成開始が早まっており、また成体においても、実験的な脱髄に対して抵抗性があり髄鞘の再形成能が亢進していることを見いだしました。分子・細胞レベルの解析から、髄鞘の形成に関わる細胞内シグナル伝達に対してPtprzが抑制的に働く仕組みも明らかになりました。これらの成果は、神経軸索の髄鞘が形成される制御メカニズムの一端を解き明かすものであり、難病である多発性硬化症等の脱髄疾患註2)に対して、髄鞘の再形成を促す新しいタイプの治療薬の開発においても大いに役立つ知見です。本研究成果は、日本時間11月8日にPLOS ONEに掲載されました。
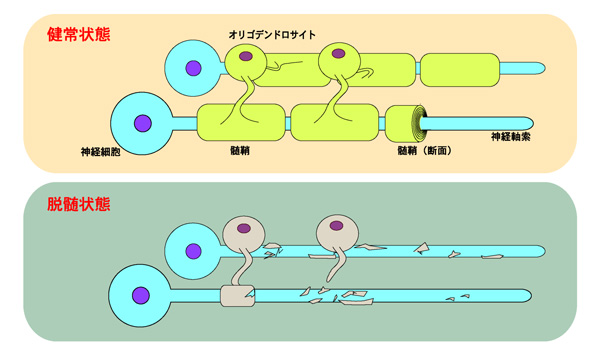
註1)図は健全な髄鞘と脱随状態を示す。オリゴデンドロサイトは神経軸索を幾重にも巻いて髄鞘を形成し、絶縁している。
註2)脱髄疾患: 多発性硬化症(MS)は主要な脱髄疾患であり、一般に炎症、脱随、神経変性を伴う中枢神経系の自己免疫疾患と考えられている。難治性疾患克服研究事業における特定疾患の一つである。平均発病年齢は30歳前後、患者数は年々増加傾向にある。四肢麻痺や失明に至ることもあり、社会生活全般を独力で行うことが困難になる。女性にやや多く発症し、結婚・出産・育児の大きな障害となる。脱髄疾患に決定的な治療法は確立されておらず、慢性的で、精神的・経済的損失、介護等による家族負担も大きい。
[研究背景]
蛋白質の特定のチロシン残基に認められる可逆的なリン酸化修飾は、細胞増殖、細胞接着・移動、細胞—細胞間のコミュニケーションといった様々な細胞機能に関わる細胞内シグナル伝達の基本的な仕組みです。この反応はリン酸基を付加するプロテインチロシンキナーゼ(PTK)と、その逆にリン酸基を除去するプロテインチロシンホスファターゼ(PTP)によって制御されます。PTKファミリーは、トランスフォーメーション(形質転換)因子として発見された経緯もあり、ガン治療薬開発における主要な創薬標的になっています。一方、PTPはPTKに約10年遅れて発見され、その役割やシグナル伝達の仕組みは今もよく判っていません。
基礎生物学研究所・統合神経生物学研究部門では、主に脳・脊髄・網膜など中枢神経系に発現しているPTPファミリー分子であるPtprzを長く研究してきました。最近、Ptprzが細胞内で脱リン酸化する基質タンパク質に、共通するモチーフ構造があることを発見し、Ptprzによって制御される細胞内シグナル伝達系に対する理解が進みました(JBC, 286, 37137-37146, 2011)。基質タンパク質の一つ、p190RhoGAPは、細胞の形や運動に関わる細胞骨格系の制御に関わっています。オリゴデンドロサイトは、その細胞膜を神経軸索に巻き付けて、髄鞘と呼ばれる絶縁体を形成し、神経伝達速度を高めるなどの役割を担っています。オリゴデンドロサイト系列の細胞において、p190RhoGAPは、PTKの一つFynキナーゼによるリン酸化によって活性化し、細胞の分化を促し髄鞘形成を誘導します。
これらの先行知見から、アクセルに相当するFynキナーゼに対して、Ptprzがブレーキのように働く役割が推測されました。そこで、既に同研究部門で作出していたPtprz遺伝子の欠失マウス(Ptprz欠損マウス)を用いて動物個体レベルでの実証研究を行いました。
[研究成果]
髄鞘の主要な構成タンパク質であるミエリン塩基性タンパク質(MBP)の発現量をマウス脳内で髄鞘形成が開始される生後10日で調べてみると、Ptprz欠損マウスではMBPの発現量が野生型マウスに較べて有意に増加していました(図2左上)。生後10日齢の脳組織を電子顕微鏡観察すると、Ptprz欠損マウスの神経繊維の髄鞘化が野生型マウスよりも早く進んでいました(図2下側:脳梁部)。初代培養細胞系を用いた解析でも、Ptprz欠損マウス由来の細胞は、オリゴデンドロサイトへより早く分化することが確かめられました。Ptprzは、神経軸索が髄鞘で覆われる発達期がくるまで、オリゴデンドロサイトを前駆細胞の状態にとどめておくストッパーのような働きを担うと考えられます(図1)。一方、成体である3ヶ月齢になると、このような差異は消失していました(図2右上)。
Ptprzの発現は発達期に限らず成体組織でも持続しています。そこで、一見変化が認められない成体マウスにおいて強制的に脱髄を誘導したときの応答性を解析しました。脱髄は、髄鞘の構成成分に由来するペプチド抗原をマウスに免疫投与して実験的自己免疫性脳脊髄炎(EAE)を誘導する手法を使いました。EAEは、多発性硬化症(MS)の動物モデルとして広く研究に用いられるものです。その結果、Ptprz欠損マウスでは、四肢麻痺など、EAEの症状が野生型マウスに較べて軽く(図3)、それに対応して脊髄組織における髄鞘損傷も軽減されていました(図4)。EAE傷害部位への炎症性細胞の浸潤の程度は野生型マウスと差異がないことから、EAEの症状の軽減は、免疫応答の差異に起因するものではないと考えられました。この結果は、成体のEAE傷害時においても、Ptprzはオリゴデンデンドロサイトの分化や髄鞘の形成/再形成に対して抑制的に働いていることを示しています。
現在、脱髄疾患の新たな治療薬として、脱髄部位に存在するオリゴデンドロサイト前駆細胞を積極的にオリゴデンドロサイトに分化・成熟させて、髄鞘の再形成を促すような薬剤の開発が求められています。本研究で明らかになったオリゴデンドロサイト分化の分子メカニズム(図1)は、Ptprzの酵素活性を選択的に阻害する化合物の有効性を示唆しています。
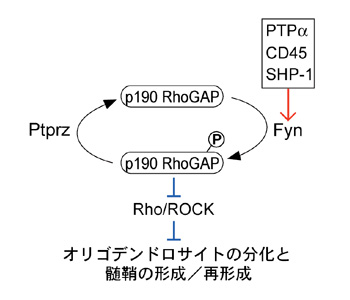
図1: オリゴデンドロサイトの分化及び髄鞘形成に関わるPtprzの役割
Ptprzは、Fynキナーゼによってリン酸化されたp190RhoGAPを脱リン酸化して、その働きを抑えることで、オリゴデンドロサイトへの分化/髄鞘の形成を抑制している。一方、PTPα、CD45、SHP-1といった他のチロシンホスファターゼ分子はFynキナーゼを活性化し、オリゴデンドロサイトの分化を促進する働きが報告されている。赤線は活性化、青線は抑制的作用を示す。
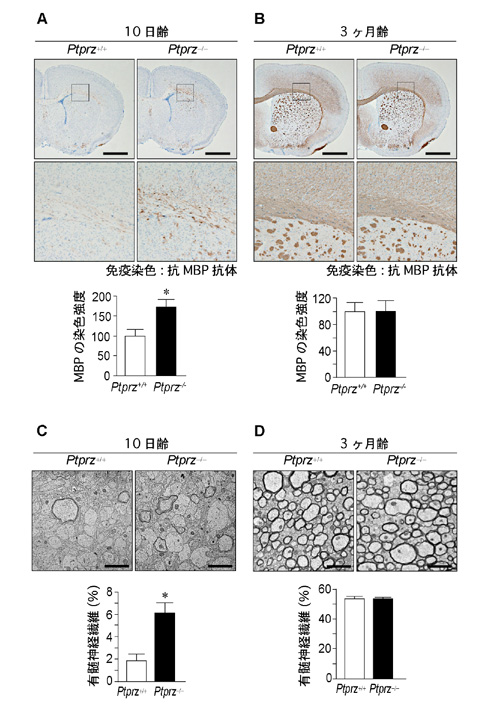
図2: 発生期のPtprz欠損マウスにおけるミエリン塩基性タンパク質の発現量及び有髄神経繊維の増加
A,B, ミエリン塩基性タンパク質(MBP)の発現解析。髄鞘形成が始まる生後10日齢(A)および成熟した3ヶ月齢(B)の野生型マウス(Ptprz+/+)及びPtprz欠損マウス(Ptprz-/-)の脳組織の抗MBP抗体染色像。スケールバー, 500 µm。下図は上図の四角エリアの拡大。下段グラフはMBP染色強度の定量評価。*P < 0.05。
C,D, 生後10日齢(C)および3ヶ月齢(D)の脳組織の電子顕微鏡観察。スケールバー, 2 µm。下段グラフは有髄神経繊維の存在比の定量評価。*P < 0.05。
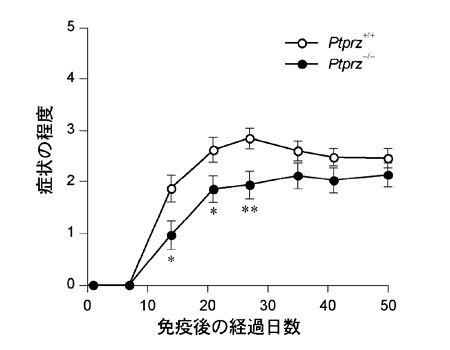
図3: EAEに対するPtprz欠損マウスの抵抗性(臨床的評価)
ミエリンオリゴデンドロサイト糖タンパク質(MOG)由来のペプチド抗原による免疫感作(0日)後の症状の経過観察。*P < 0.05, **P < 0.01。
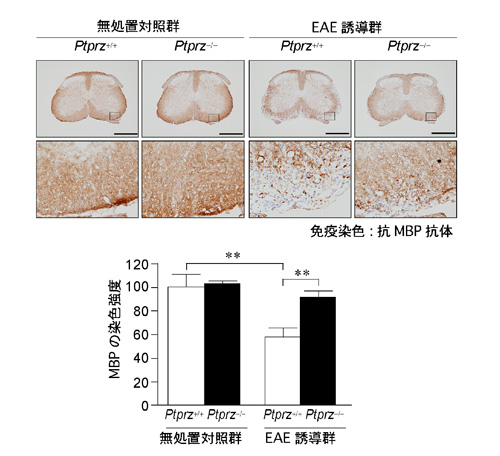
図4: EAEに対するPtprz欠損マウスの抵抗性(組織学的評価)
抗MBP抗体を用いたMOGペプチド抗原による免疫感作35日後(EAE誘導群)と無処置対照群の脊髄組織のMBPの免疫染色解析。スケールバー 500 µm。下図は上図の四角エリアの拡大。下段グラフはMBP染色強度の定量評価。Ptprz欠損マウスではMBPがほとんど失われていない。**P < 0.01。
[成果の意義]
発達期の脳神経系においてPtprzが関わる髄鞘が形成される仕組みが明らかになりました。脱髄疾患に対する新薬開発においてPtprzは有望な標的分子の一つになると考えられます。
[発表雑誌]
科学雑誌 PLOS ONE 2012年11月7日 公開(日本時間11月8日午前4時)
論文タイトル:Protein Tyrosine Phosphatase Receptor Type Z Negatively Regulates Oligodendrocyte Differentiation and Myelination
著者:Kazuya Kuboyama, Akihiro Fujikawa, Makoto Masumura, Ryoko Suzuki, Masahito Matsumoto, Masaharu Noda
[研究グループ]
本研究は、基礎生物学研究所・統合神経生物学研究部門とアスビオファーマ株式会社との共同研究として実施されました。
[研究サポート]
本研究の一部は、文部科学省科学研究費補助金のサポートを受けて行われました。
[本件に関するお問い合わせ先]
基礎生物学研究所 統合神経生物学研究部門
教授: 野田 昌晴 (ノダ マサハル)
TEL: 0564-59-5846(研究室)
E-mail: madon@nibb.ac.jp
URL: http://niwww3.nibb.ac.jp/
[報道担当]
基礎生物学研究所 広報室
TEL: 0564-55-7628 FAX: 0564-55-7597
E-mail: press@nibb.ac.jp