10.1 Insertion of GUS (GFP, YFP) genes to form a
fusion protein with a target gene
Tomomichi Fujita and Yuji Hiwatashi
1. Strategy
A reporter gene to fuse with a native gene is knocked-in with gene
targeting technique. Spatiotemporal regulation of the fusion protein driven by
a native promoter can be examined. However, the stability and localization of
the fusion protein may be different form the native protein. Especially when
similar phenotypes to disruptants are observed in the transformants with the fusion
protein, the fusion protein likely have different function, such as dominant
negative function, from the native protein. Technique on histochemical immuno
staining has long been expected, but is applicable only in protonemata, but not
in gametophores and sporophytes.
2.
Construction and transformation
The uidA (GUS), GFP,
and modified YFP genes are used as reporter genes. The reporter gene is fused to either C-terminal or N-terminal
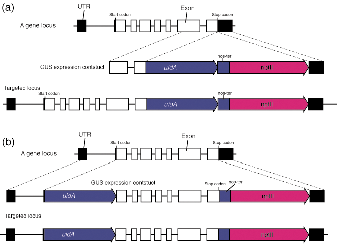
end of the targeted gene (Fig. 1). Comparison of localization between
C-terminal and N-terminal fusion proteins will give implications for the
reliability of the localization, and both constructs should be designed.
[C-terminal
fusion]
・ GUS
+ G418 (nptII) :pGUSmutNPTII, pTN83, pTN84, pTN85
・ GFP
+ G418 (nptII):pGFPmutNPTII
・ YFP
+ G418 (nptII):pCtrnNPTII2
・ GFP
+ Hygromycin (hyg) :pYRG2
・ GFP
+ Zeocin (zeocin):pHIZ2
[N-terminal
fusion]
・ GFP
+ Zeocin (zeocin):pHIZ3
・ YFP
+ G418 (nptII):pCtrnNPTII2
Figure 1. Maps for constructions of C-terminal fusion
(a) and N-terminal fusion (b)
i)
C-terminal GUS(GFP,
YFP)fusion
construction (Fig. 1a)
A more than
1 kb genomic DNA fragment of a targeted gene whose 3' end is just before its
stop codon is inserted in 5' multicloning site of the vector (Fig. 1a). A
method to obtain the genomic sequences are shown in 10.0.
The uidA gene becomes the same reading frame as the
targeted gene. Another more than 1 kb genomic DNA fragment whose 5' end is
the stop codon of the targeted gene is inserted in 3' multicloning site of
the vector (Fig. 1a). The size of the homologous genomic DNA fragments can
be reduced to 500 bp or less, although the rate of homologous recombination
becomes worse.
(Example)
1.
Amplify a genomic fragment
for the 5' end as mentioned above by PCR. Add restriction sites at the 5' end
of primers for cloning. Select restriction sites that do not cut the genomic
fragment.
4.
Linearize the plasmid by
restriction enzymes to excise the DNA fragment for targeting from a vector and
transform protoplasts.
ii) N-terminal
GUS(GFP,
YFP)fusion
construction (Fig. 1b)
Three DNA
fragments are inserted into a cloning vector (Fig. 1b). At first, a more than 1
kb genomic DNA fragment of a targeted gene whose 3' end is the start ATG is inserted
into 5' end of a reporter gene without its start codon. Secondly, a genomic DNA
fragment covering a coding region of the targeted gene that starts just after
its start codon and that ends at its stop codon is inserted into 3' end of a
reporter gene to make a translational fusion product. Finally, a more than 1 kb
genomic DNA fragment that starts just after the stop codon of the targeted gene
is inserted into 3' end of the nptII cassette.
(Example using
pHIZ3)
1. Amplify a
genomic fragment of 5' untranslated region as mentioned above by PCR. Add
restriction sites at the 5' end of primers for cloning. Select restriction
sites that do not cut the genomic fragment. After cloning the fragment, confirm
the correct insertion and no PCR errors by sequencing, especially for the
reading frame.
2. Amplify a
genomic fragment as mentioned above by PCR with primers carrying SalI
and ClaI sites. After cloning the fragment, confirm the correct insertion
and no PCR errors, especially for the reading frame.
3. Amplify a genomic
fragment of 3' untranslated region by PCR with primers having restriction sites
for the cloning. Select restriction sites that do not cut the genomic fragment.
After cloning the fragment, confirm the correct
insertion and no PCR errors by sequencing, especially for the reading frame.
4. Linearize
the plasmid by restriction enzymes to excise the DNA fragment for targeting
from a vector and transform protoplasts.
3. Selection
of transformants with a correct insertion
PCR and
Southern blot analyses are used to select transformed lines with a correct
insertion. PCR is used at the first stage of screening, and then southern
analysis is performed. To avoid an unexpected insertion, You definitely need to
perform southern hybridization to proceed to further analyses of the
transformants.
(Example
for 3' knock-in)
(1) PCR
analyses
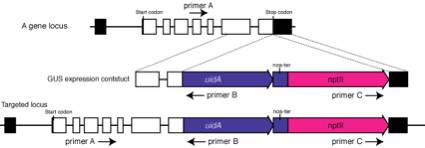
1.
Extract a genomic DNA from the candidate stable transformants.
2.
Perform two kinds of PCRs:
(first PCR) Perform
PCR using primer A and B. Primer A needs to locate at 5' region to the expected
5' recombination site and does not locate in the targeted fragment.
(second PCR)
Perform PCR using primer B and C. No PCR products will be obtained. If products
are amplified, targeting fragments are likely knocked in as a tandem repeat.
The tandem insertions often happens in our experimental conditions. To prevent
PCR error, confirmation by southern hybridization is necessary.
(2) Southern
blot analyses
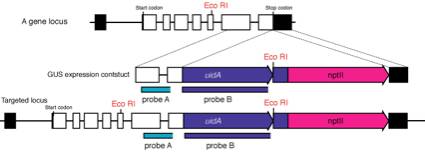
1.
Extract genomic DNA by CTAB method from the candidate
transformants selected by PCR.
2.
Digest genomic DNA with restriction enzymes (ex. EcoRI
in the figure) and blot to a nylone membrane.
3.
We usually use two kinds of probes: one locates in genomic region
(probe A) and another in uidA to prevent the unexpected insertions that
sometimes happen!
Appendix
A targeting
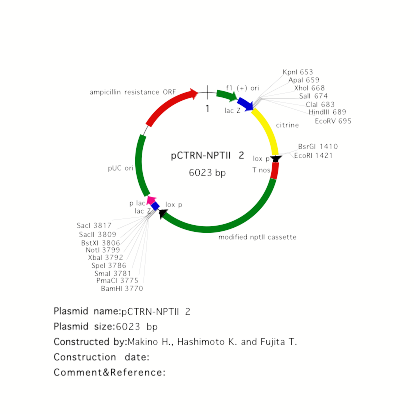
vector for YFP fusion: pCTRN-NPTII 2
pCTRN-NPTII 2
carries a modified YFP, called citrine, as a reporter. Citrine shows more
decreased pH and halide sensitivity than YFP (Griesbeck et al. JBC 276, 2001).
Citrine was kindly provided by Dr. R. Tsien. Place a genomic fragment
containing a coding region into EcoRV site to make
C-terminal citrine fusion. For N-terminal citrine fusion, BsrGI site is
available.
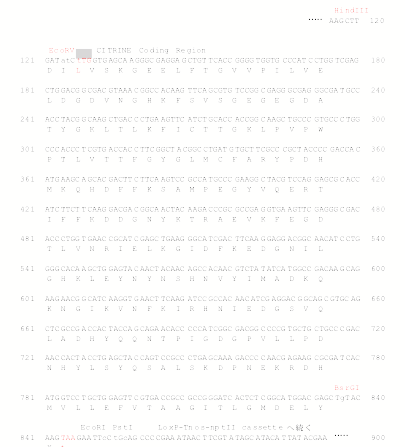
Sequence
around citrine gene in pCTRN-NPTII 2 (See below)