10.1ü@
Expression and localization of a fusion protein of a
targeted gene and a reporter gene
Yuji Hiwatashi, Tomomichi
Fujita, and Mitsuyasu Hasebe
There
are three methods to analyze spatiotemporal expression patterns and
localization of protein. In situ hybridization
of mRNA and immunostaining of gametophore and
sporophyte tissue in P. patens are
not successful in our hands. We observe a protein fused with a reporter
protein. The following three methods are introduced in this chapter: (1)
Knock-in a reporter gene to form a fusion protein with a targeted gene; (2)
Constitutive expression of a fusion protein of a targeted protein and a
reporter protein, which is targeted at a neutral site; (3) Transient expression
of a fusion protein of a targeted protein and a reporter protein in
protoplasts. The best way is the first one but the latter two methods are
easier and useful only for preliminary analyses.
(1) Insertion of a reporter gene to form a
fusion protein with a targeted gene
1. Strategy
A
reporter gene is knocked-in with the gene targeting technique to fuse with a
targeted gene. Spatiotemporal localization of the fusion protein driven by a
native promoter can be examined. However, the stability and localization of the
fusion protein may be different from those of the native targeted protein. We
always need to confirm the phenotypes of knocked-in lines are not distinguished
from those of wild type plants. Histochemical immunostaining is ideal to
confirm the protein localization detected by the fusion protein. However, we
can routinely perform immunostaining of protonemata, but it is still
challenging to immunostain gametophore and sporophyte
cells.
2. Construction and transformation
The uidA (GUS) (Jefferson
et al., 1987),
Cerulean (modified CFP) (Rizzo
et al., 2004),
sGFP (modified GFP) (Chiu
et al., 1996),
Citrine (modified YFP) (Griesbeck et al., 2001),
and tagRFP (modified RFP) (Merzlyak et al., 2007) genes have been used as reporter genes in our laboratories. The reporter gene is fused to either the
C-terminal or N-terminal end of the targeted gene (Fig. 1).
Figure 1.ü@
Maps for constructions of C-terminal fusion (a) and N-terminal fusion
(b)
i) C-terminal reporter
protein fusion construction (Fig. 1a)
A
3üf DNA fragment of a targeted gene (>1 kb long for effective homologous
recombination) just before its stop codon is inserted in the 5' multicloning site of the vector (see a map of each vector
and Fig. 1a) so that the uidA gene is
fused in-frame with the targeted gene. Another DNA fragment (>1 kb long for
effective homologous recombination) whose 5' end is the stop codon of the
targeted gene is inserted in the 3' multicloning site
of the vector (see a map of each vector and Fig. 1a). The size of the
homologous genomic DNA fragments can be reduced to less than 1 kb, although the rate of homologous recombination becomes lower.
(Example)
1. ü@Amplify a genomic fragment for the 5' end by
PCR. Include restriction sites at the 5' end of the primers for cloning. Select
restriction sites that do not cut the genomic fragment.
4. ü@Cut the plasmid by restriction enzymes to
excise the targeting fragment from the vector and transform protoplasts.
[C-terminal
fusion] (Table 1)
üE GUS: pTN85 (G418 resistance)
üE Modified CFP (Cerulean): pCerulean-NPTII
(G418 resistance)
üE Modified GFP (sGFP): pGFPmutNPTII (G418
resistance), pYHG2 (Hygromycin resistance), pHIZ2 (Zeocin resistance)
üE Modified YFP (Citrine): pCTRN-NPTII2 (G418 resistance), pCit-aphIV (Hygromycin resistance)
üE Modified RFP (TagRFP): pTagRFP-NPTII
(G418 resistance)
Table 1. Available plasmids for C-terminal
reporter fusion
|
Name |
Reporter protein |
Resistant (P. patens) |
Resistant (E. coli) |
Removal of a nos terminator and an antibiotics-resistant marker
cassette by Cre recombinase |
Reference, Accession
no. |
|
pTN85 |
GUS |
G418 |
Ampicillin |
Yes |
(Sakakibara et al., 2008), AB267707 |
|
pCerulean-NPTII |
Cerulean |
G418 |
Ampicillin |
Yes |
|
|
pGFPmutNPTII |
sGFP |
G418 |
Ampicillin |
No |
(Hiwatashi
et al., 2008) |
|
pYRG2 |
sGFP |
Hygromycin |
Ampicillin |
No |
|
|
pHIZ2 |
sGFP |
Zeocin |
Ampicillin |
No |
|
|
pCTRN-NPTII2 |
Citrine |
G418 |
Ampicillin |
Yes |
|
|
pCit-aphIV |
Citrine |
Hygromycin |
Ampicillin |
Yes |
|
|
pTagRFP-NPTII |
TagRFP |
G418 |
Ampicillin |
Yes |
|
ii) N-terminal reporter protein fusion
construction (Fig. 1b)
Three
DNA fragments are inserted into the cloning vector (Fig. 1b). At first, approximately
1 kb genomic DNA fragment of the targeted gene whose
3' end is the start ATG is inserted into the 5' end of a reporter gene. Secondly,
a genomic DNA fragment covering the entire coding region of the targeted gene (from
the second codon just after its putative start codon to the putative stop codon)
is inserted at the 3' end of the reporter gene to make a translational fusion
product. Finally, an approximately 1 kb genomic DNA fragment
that starts after the stop codon of the targeted gene is inserted into 3' end
of the nptII cassette.
(Example 1 using pHIZ3)
1.
Amplify a genomic fragment of 5' untranslated region as mentioned above by PCR.
Add restriction sites at the 5' end of the primers for cloning. Select
restriction sites that do not cut the genomic fragment. After cloning the
fragment, confirm the correct insertion, absence of PCR errors and
reading-frame by sequencing.
2.
Amplify a genomic fragment as described above by PCR with primers carrying SalI and ClaI sites. After
cloning the fragment, confirm the correct insertion, absence of PCR errors and
reading-frame by sequencing.
3. Amplify
a genomic fragment of 3' flanking sequence by PCR with primers having
restriction sites for the cloning. Select restriction sites that do not cut the
genomic fragment. After cloning the fragment, confirm
the correct insertion and absence of PCR errors by sequencing.
4.
Linearize the plasmid by restriction enzymes to excise the targeting fragment from
the vector and transform protoplasts.
(Example 2 using pCTRN-NPTII2)
1.
Add a start codon, ATG at the 5' end of the reverse primer because the start
codon of Citrine in pCTRN-NPTII2 is
replaced with TTG. Phosphorylate this reverse primer with T4 polynucleotide
kinase before use in PCR. Add restriction sites at the 5' end of the forward primer
for cloning. Select restriction sites that do not cut the genomic fragment. Amplify
a genomic fragment of 5' untranslated region as mentioned above by PCR. Digest
the PCR fragment with the restriction enzyme that recognizes the restriction
site in the forward primer and clone the PCR fragments between the EcoRV and the
restriction site of pCTRN-NPTII2. After cloning the fragment, confirm the correct
insertion, absence of PCR errors and reading-frame by sequencing.
2.
Amplify a genomic fragment as described above by PCR with primers carrying Acc65I, BsiWI, or BsrGI site and clone the fragment
in the BsrGI
site of pCTRN-NPTII2. After cloning the fragment, confirm the correct insertion,
absence of PCR errors and reading-frame by sequencing.
3. Amplify
a genomic fragment of 3' flanking sequence by PCR with primers having
restriction sites for the cloning. Select restriction sites that do not cut the
genomic fragment. After cloning the fragment, confirm
the correct insertion and absence of PCR errors by sequencing.
4.
Linearize the plasmid by restriction enzymes to excise the targeting fragment from
the vector and transform protoplasts.
ü@[N-terminal fusion] (Table 2)
üE Modified GFP (sGFP): pHIZ3 (Zeocin resistance)
üE Modified YFP (Citrine): pCTRN-NPTII2 (G418 resistance)
Table 2. Available plasmids for N-terminal
reporter fusion
|
Name |
Reporter protein |
Resistant (P.patens) |
Resistant (E.coli) |
Removal of a nos terminator and an antibiotics-resistant marker
cassette by Cre recombinase |
Reference, Accession
no. |
|
pHIZ3 |
sGFP |
Zeocin |
Ampicillin |
No |
For in-frame fusion, SalI and ClaI sites are available. |
|
pCTRN-NPTII2 |
Citrine |
G418 |
Ampicillin |
Yes |
For in-frame fusion, a BsrGI site are
available. Start codon (ATG) of Citrine is replaced with TTG. You need
to add a start codon just before coding sequence of Citrine in-frame. |
3. Selection of transformants with a correct
insertion
PCR
and Southern blot analyses are used to select transformed lines with a correct
insertion. PCR is used at the first stage of screening, and then southern
analysis is performed. To avoid an unexpected insertion, You
definitely need to perform southern hybridization before proceeding to further
analyses of the transformants.
(Example for 3' knock-in)
(1) PCR analyses
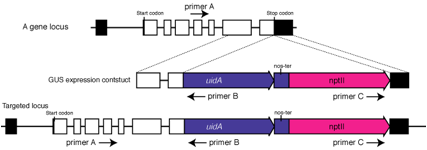
1. Extract
a genomic DNA from the candidate stable transformants.
2. Perform
two kinds of PCRs:
(First
PCR) Perform PCR using primers A and B. Primer A needs to locate in a region 5üf
to the expected 5' recombination site and be located external to the targeting
sequence.
(Second
PCR) Perform PCR using primers B and C. No PCR products should be obtained. If
products are amplified, the targeting fragments are probably knocked in as a
tandem repeat. Tandem insertions often happen under our experimental
conditions. To prevent PCR error, confirmation by southern hybridization is
necessary.
(2) Southern blot analyses
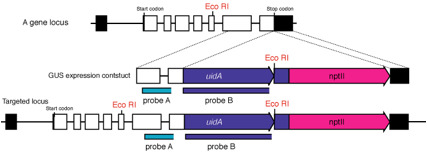
1. Extract
genomic DNA by CTAB method from the candidate transformants selected by PCR.
2. Digest
genomic DNA with restriction enzymes (ex. EcoRI in the figure) and blot to
a nylon membrane.
3. We
usually use two kinds of probes: one locates in genomic region (probe A) and
another in uidA to identify the
unexpected random insertions that sometimes happen!
3. Removal of a nos
terminator and an antibiotics-resistant marker cassette by loxP
and Cre recombinase
Several
vectors contain the loxP sites where are located
before a nos terminator and after an
antibiotic-resistant marker cassette (Fig. 2). These DNA region flanked by the loxP sites can be removed with Cre
recombinase.ü@
Figure 2. Schematics of pTN85
 Example for Removal of
the resistant marker cassette
Example for Removal of
the resistant marker cassette
A
linearized selection marker cassette ligated with a vector fragment is
co-introduced and functions as an episome (Muren et al., 2009). Episomal transformants obtained with
linearized DNA contain more episomal plasmids than
transformants obtained with circular DNA (Muren et al., 2009). An excess amount of Cre recombinase may be harmful to P. patens genome stability
and a circular selection marker cassette is used.
1.
A circular plasmid of pTN75 containing Cre recombinase expression and hygromycin resistant cassettes
is introduced into transformant lines. The vector information is in the PHSYCObase. When we cut plasmids with a restriction enzyme,
the rate of excision greatly decreased.
2.
Transformed protoplasts are cultivated for 4 days on PRM/B medium without
hygromycin, and then for 2 weeks on BCDAT medium with hygromycin.
3.
We transferred 24 colonies to BCDAT medium without hygromycin and cultivated
them for 2 weeks, during which the circular plasmids transiently incorporated
in P. patens cell are removed. After 2 weeks of culture, protonema tissue grown
after moving to non-selection medium is transplanted to new medium for further
culture.
4.
We examined deletion of the selection marker cassette with Green-PCR and
removal of the Cre recombinase
vector by inoculating on BCDAT medium containing hygromycin.
References
Chiu,
W., Niwa, Y., Zeng, W.,
Hirano, T., Kobayashi, H., and Sheen, J. (1996).
Engineered GFP as a vital reporter in plants. Curr. Biol. 6: 325-330.
Griesbeck, O., Baird, G.S., Campbell, R.E., Zacharias, D.A., and Tsien, R.Y. (2001).
Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J. Biol. Chem. 276: 29188-29194.
Hiwatashi, Y., Obara, M., Sato, Y.,
Fujita, T., Murata, T., and Hasebe, M. (2008).
Kinesins are indispensable for interdigitation of phragmoplast microtubules in
the moss Physcomitrella patens. Plant Cell 20:
3094-3106.
Jefferson,
R.A., Kavanagh, T.A., and Bevan, M.W. (1987). Gus Fusions - Beta-Glucuronidase
as a Sensitive and Versatile Gene Fusion Marker in Higher-Plants. Embo Journal 6: 3901-3907.
Merzlyak, E.M., Goedhart,
J., Shcherbo, D., Bulina,
M.E., Shcheglov, A.S., Fradkov,
A.F., Gaintzeva, A., Lukyanov,
K.A., Lukyanov, S., Gadella,
T.W., and Chudakov, D.M. (2007). Bright monomeric red fluorescent
protein with an extended fluorescence lifetime. Nat Methods 4: 555-557.
Muren, E., Nilsson, A., Ulfstedt, M.,
Johansson, M., and Ronne, H.
(2009). Rescue and characterization of episomally
replicating DNA from the moss Physcomitrella. Proc. Natl. Acad. Sci. USA
106: 19444-19449.
Rizzo,
M.A., Springer, G.H., Granada, B., and Piston, D.W. (2004). An improved cyan fluorescent protein
variant useful for FRET. Nat Biotechnol 22: 445-449.
Sakakibara, K., Nishiyama, T., Deguchi,
H., and Hasebe, M.
(2008). Class 1 KNOX
genes are not involved in shoot development in the moss Physcomitrella patens
but do function in sporophyte development. Evol Dev 10: 555-566.
(2) Constitutive expression of a fusion protein
of a targeted protein and a reporter protein, which is targeted at a neutral
site
(A) Strategy
ü@
cDNA-GFP fusion constructs are
targeted to a certain ügplatform lociüh and stably overexpressed. Localization of
the fusion proteins are then examined. We use the Pphb7,
PpMADS2, or BS213 loci as platforms.ü@
(B) Available constructs using
gateway system
Original
gateway constructs were kindly provided from Dr. Tsuyoshi Nakagawa (Shimane
University, Japan).
For
stable transformation to BS213 targetingü@
(Ref. for BS213 locus,ü@ Schaefer et al. 1997, Plant J.)
[C-mRFP fusion/35S promoter]
BS213 5'-35S-R1-Cm-ccdB-R2-mRFP-Tnos-Zeo selection cassette-BS213 3'ü@
[N-mRFP fusion/35S promoter]
BS213 5'-35S-mRFP-R1-Cm-ccdB-R2-Tnos-Zeo selection cassette-BS213 3'ü@
[C-mRFP fusion/7113 promoter]$
BS213 5'-7113-R1-Cm-ccdB-R2-mRFP-Tnos-Zeo selection cassette-BS213 3'ü@
[N-mRFP fusion/7113 promoter]
BS213 5'-7113-mRFP-R1-Cm-ccdB-R2-Tnos-Zeo selection cassette-BS213 3'ü@
(3) Transient expression
of a fusion protein of a targeted protein and a reporter protein in protoplasts
(A) Strategy
ü@
cDNA-GFP fusion constructs are
introduced into protoplasts and TRANSIENTLY expressed using a weak 35S promoter
in the regenerated cells. Multi-copy cDNA-GFP constructs are usually introduced
to protoplasts in our hands, which can cause mis-localization
of the fusion protein because of its excess amount. The transient assay is a
much more convenient and rapid method than that with stable transformants in
(1), and a method to introduce a smaller number of plasmids should be
established in future.
A
rescue of a knockout phenotype by cDNA-GFP expression proves the functionality
of the fusion protein, although the rescue does not necessarily give support
for the localization of cDNA-GFP fusion protein.
(B) Available constructs using
gateway system
Original
gateway constructs were kindly provided from Dr. Tsuyoshi Nakagawa (Shimane
University, Japan)
35S-R1-Cm-ccdB-R2-sGFP-Tnosü@ü@ü@ü@ü@ [C-GFP
fusion]
35S-sGFP-R1-Cm-ccdB-R2-Tnosü@ü@ü@ü@ü@ [N-GFP
fusion]
35S-R1-Cm-ccdB-R2-mRFP-Tnosü@ü@ü@ü@ü@ [C-RFP
fusion]
35S-mRFP-R1-Cm-ccdB-R2-Tnosü@ü@ü@ü@ü@ [N-RFP
fusion]
35S-R1-Cm-ccdB-R2-YFP-Tnosü@ü@ü@ü@ü@ü@ [C-YFP
fusion]
35S-YFP-R1-Cm-ccdB-R2-Tnosü@ü@ü@ü@ü@ü@ [N-YFP
fusion]
The
CaMV-derived 7113 promoter is stronger than the 35S
promoter in P. patens.ü@ Drs. I. Mitsuhara
and Y. Ohashi kindly provided the promoter (Mitsuhara et al., Plant Cell Physiol.ü@ 1996).
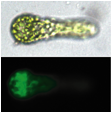
(Figure)
A cDNA1-GFP fusion plasmid was transiently expressed in protoplasts and in a
3-cell stage, asymmetric protein localization in a basal cell was observed.