| DIVISION OF GENE EXPRESSION AND REGULATION II | ||
|
||
* to Center for Trangenic Animals and Plants, NIBB (Sep, 1, 2003) ** from Sep. 1, 2003 *** from Nov. 30, 2003 |
||
The genomes of higher organisms contain
significant amounts of repetitive sequences, which in general, are
unstable. At present, neither the physiological function(s) of repeated
sequences nor the mechanism controlling the instability is fully understood.
To clarify these aspects, we are pursuing the following themes using
E. coli and S. cerevisiae: (1) the amplification
mechanism of repeated sequences or genes, especially rRNA repeated
genes, (2) the mechanism of replication fork block-dependent recombination,
a key reaction that increases or decreases the number of repeats,
and (3) development of in vivo artificial gene amplification systems.
Structural and functional analyses of the E. coli genome
are also being carried out. In 2003, work on the following three subjects
has advanced our knowledge of the dynamics and structure of the genome.
The DNA replication fork blocking sites are found in genomes of various
organisms. In E. coli, the fork block site, called Ter,
has the fork blocking activity in a polar fashion. For the blocking
activity, two components are required, one is a cis-element, Ter,
the other is the Ter-binding protein, called Tus protein. In eucaryotes,
the similar blocking site, called RFB (replication fork blocking barrier),
has been identified in the ribosome RNA gene (rDNA) cluster from yeast
to human. In S. cerevisiae, about 150 copies of rDNA are
clustered in a specific region on the chromosome XII. The RFB is an
approximately 100-bp DNA sequence located near the 3' end of the rRNA
genes in the yeast Saccharomyces cerevisiae. This site inhibits
the progression of the DNA replication fork coming from the direction
opposite to 35S rDNA transcription. However, the RFB-binding protein
has not been identified so far. The most likely candidate for the
RFB-binding protein is Fob1p, because it had shown to be required
for the replication fork blocking activity at the RFB site. Furthermore,
it was found later that it is essential for recombination in the ribosomal
DNA (rDNA), including increase and decrease of rDNA repeat copy number,
production of extra-chromosomal rDNA circles, and possibly homogenization
of the repeats. Despite the central role that Foblp plays in both
replication fork blocking and rDNA recombination, not only the molecular
mechanism by which Fob1p mediates these activities, but also its RFB-binding
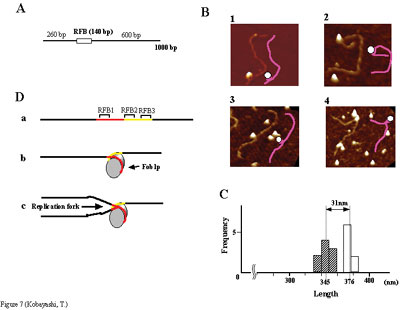
activity had not been determined. We have shown by using chromatin
immuno-precipitation, gel shift, foot-printing, and atomic force microscopy
assays that Fob1p directly binds to the RFB (see Figure A and B).
Fob1p binds to two separated sequences in the RFB (Figure D). A predicted
zinc finger motif in Fob1p was shown to be essential for the RFB binding,
replication fork blocking, and rDNA recombination activities. The
RFB seems to wrap around Fob1p, and this wrapping structure may be
important for function in the rDNA repeats (Figure B, C and D). Recombination hot-spots are DNA sequences which enhance recombination
around the region. HOT1 is one of the well-studied recombination
hot-spot in mitotic yeast cells. Because HOT1 includes a
transcription promoter sequence of RNA polymerase I (PolI) which is
responsible for the 35S ribosomal RNA gene (rDNA) transcription, and
the HOT1 activity is abolished in a PolI defective mutant,
transcription of HOT1 was thought to be an important factor
for the recombination stimulation. However, it is not clear whether
the transcription itself or other phenotypes shown in the PolI mutant
activates the recombination. To understand the role of transcription,
we used a highly-activated PolI transcription system for HOT1
in a strain whose rDNA were deleted (rdnDD).
In the rdnDD cells, it is known
that the nucleolus disappears and PolI, which is normally localized
to the nucleolus, diffuses into the cytoplasm. In the rdnDD
strain, the HOT1 transcription was increased by about 25
times as compared with the wild type. Recombination activity stimulated
by HOT1 was also elevated by about 15 times as compared with
that of wild type. These results indicate that frequency of PolI transcription
in HOT1 determines efficiency of the recombination. Moreover,
Fob1p, which is essential for both HOT1 recombination and
transcription activities, was dispensable in the rdnDD
strains, thereby suggesting that the protein may be functioning as
a PolI transcription activator in the wild type strain. Gene amplification is a phenomenon widely found in the genomes of
various organisms and probably has played an important role in the
processes of gene evolution. For example, rDNA must have amplified
during evolution and in some amphibians, further amplification occurs
during their development. Oncogene amplification is frequently observed
when cancer is in progress. Gene amplification also occurs when cultured
cells acquire drug-resistance. However, human cultured cells lack
the ability to amplify drug-resistance genes at a detectable level.
Spontaneous amplification also occurs in insects and plants, but the
molecular mechanism remain uncertain. Thus, if a novel artificial
amplification could be realized using normal organisms, it would be
helpful for better understanding the basic mechanism of the various
amplifications. Thus, we have tried to develop a novel designed amplification
system using a normal S. cerevisiae strain. We assumed that
a mode of replication, named double rolling-circle replication (DRCR),
could amplify a gene between two directed replication forks, and tested
it on plasmid-derived mini-chromosomes or on a resident chromosome
utilizing break-induced replication (BIR). The latter system produced
three kinds of amplification products; type-1 containing 5~7 copies
of an amplification marker, leu2d, with the expected structure,
type-2 containing several dozen copies of leu2d with a similar
structure to type-1, but the sequences flanked by the two inverted
leu2ds are oriented randomly, and type-3 having an acentric multi-copy
mini-chromosome with leu2ds. The type-2 and -3 products seem
to correspond to HSR (homogeneously staining region) and DM (double
minute) in higher eukaryotes. Surprisingly, the two latter types were
also generated without HO cleavage, though at low frequency. This
system may provide insights into the molecular mechanism of gene amplification.
Escherichia coli is one of the organisms that has been most extensively analyzed physiologically, biochemically and genetically. Of all E. coli strains, E. coli K12 W3110 has probably been used most frequently as the wild-type strain in these experiments. Recently, we determined the complete nucleotide sequence of the genome of strain W3110, mainly by sing lambda phages from Kohara’s bank. Previously a US group determined the genomic sequence from another K12 wild-type derivative, MG1655. Both strains were derived from a common ancestor strain, W1485, probably about 50 years ago. Comparing the two sequences, we obtained the following results. The total number of nucleotides in the W3110 genome is 4660170 bp. There were 349 bp conflicts between the sequences of W3110 and MG1655. Re-sequencing of the conflict sites by a PCR method using genomic DNA as templates revealed that only eight sites (9 bp) were true conflicts. Seven of them are base-change type conflicts and one is a two base frame-shift. All of these differences reside within genes, seven in ORFs and one in a 23S rRNA gene. We attended the E. coli K12 annotation workshop held at Woods
Hole Marine Biology Research Institute, Mass., USA, 2003, Nov. 13-18,
which was organized by Dr. Monica Riley. In this workshop, we discussed
(1) the definition of the starting point of the E. coli genome
sequence, (2) boundaries of ORFs, that is start and end points of
ORFs, and (3) the descriptions of gene names and their function. We
agreed there to make an effort to complete a new annotation version
using our revised sequence by the coming February. We can safely say
that the revised sequence and the annotation will be among the most
accurate of all the genomes whose sequences have been determined.
|
||
| Publication List:
Takeuchi, Y., Horiuchi, T., & Kobayashi, T. (2003) Transcription-dependent recombination and the role of fork collision in yeast rDNA. Genes Dev. 17, 1497-1506 Higuchi K, Katayama T, Iwai S, Hidaka M, Horiuchi T, Maki H.(2003). Fate of DNA replication fork encountering a single DNA lesion during oriC plasmid DNA replication in vitro. Genes Cells 8, 437-49 Kobayashi, T. (2003) The replication fork barrier site forms a unique structure with Fob1p and inhibits the replication fork. Mol. Cell. Biol. 23, 9178-88. |