9.
How
to transform Physcomitrella patens
9.1
PEG-mediated
transformation
ü@
Yuji Hiwatashi and Mitsuyasu
Hasebe
Transformation of Physcomitrella patens has been performed with the polyethylene glycol (PEG)-mediated DNA introduction to protoplasts, with the particle bombardment to live cells (see 9.2), and with the agrobacterium-mediated DNA introduction to protonemata or protoplasts (see 9.5). The PEG-mediated DNA transfer method is the most efficient and feasible for homologous recombination mediated-gene targeting. DNA bombardment usually provides unstable transformants without recombination into Physcomitrella genome and is mainly used for transient assay. Agrobacterium-mediated method is used for a specific purpose; such as high-throughput screening (see 9.5).
1. Preparation
of protonemata for transformation
(1)
Inoculate a piece of protonemata with tweezers to BCDATG medium and cultivate
for three weeks. The original protonemata are usually composed of chloronemata
and caulonemata. We will make chloronema-rich protonemata by the following
procedures, because transformation rates of chloronemata are higher than those
of caulonemata.
(2)
After each inoculated protonemata forms a colony after three weeks, move three
colonies to a test tube with 25 mL sterile water and chopped with a Polytron homogenizer
(Kinematica, AG, Lucerne, Switzerland) for 10 sec at the minimum speed of the
Polytron PT2100 with a DA2120/2 generator shaft or at speed level 4 for the
Polytron model K with a PTA20TS generator shaft.
(3)
Spread 2.5 ml of the solution of chopped protonemata on a pretreated cellophane
sheet layered on BCDATG medium (see 2.2) in a 9-cm Petri dish. Incubate the
inoculated Petri dish at 25üŗC under continuous white light (at an intensity of
30-80 µmol m-2 s-1) for 7 days without sealing the
plate.
(4) Collect all protonemata on the plate,
chop with the above conditions, spread, and cultivate as (3) for three times
and you can get chloronema-rich protonemata.
(5) Collect the chloronema-rich
protonemata of (4), chop with the above conditions, spread, and cultivate under
the same conditions for 3 to 5 days instead of 7 days. The regeneration
rate of protoplasts is critical for transformation efficiency. Protonemata
grown for 3 to 5 days after spreading should be used for protoplast isolation.
Older protonemata cause dramatically low transformation efficiency.
Protonemata cultivated on a cellophane-overlaid plate for 5
days after chopping

2. Plasmid
preparation
1. Culture E. coli harboring plasmid DNA.
2. Prepare
plasmids using QIAGEN Hi-Speed Plasmid Midi Kit or Promega Wizard Mini-prep Kit
or other similar kit.
3. Digest the
plasmid DNA with an appropriate restriction enzyme to linearize.
4. Purify the
digested plasmid with phenol/chloroform extraction and precipitate by adding 1/10th
the volume of 3M NaOAc (pH5.2) and 2 volumes of ethanol.
5. Dissolve
the precipitated plasmid with TE, adjusting at the concentration of 0.2~1.0
µg/µl.
6. Store at
–30˚C.
*
If you use PCR amplified DNA fragments, PCR products should be ethanol-precipitated
with 1/10 volume of 3M NaOAc (pH5.2) and resuspend in TE.
3.
Transformation
1. (Day 1) Culture
protonemata after chopping. Grow them at 25˚C for 4 days (or 3 to 5 days).
2. (Day 4) Chop
propagated protonemata again and grow them at 25˚C for 4 days on a new
plate.
3. (Day 8~9)
Transformation followed by incubation of transformed protoplasts in a top agar
for 3 to 5 days.
4. (Day 12)
Transfer the top agar containing regenerating protoplasts onto a selection
plate supplemented with antibiotics. Cultivate for more than 3 weeks on the
selection medium.
5. (Day 43; 3
weeks after TF) Transfer a fragment of a colony grown onto the 1st selection
plate to an antibiotic-free plate. Incubate for more than one week.
6. (Day 50; 4
week after TF) Transfer a fragment of a colony grown on the non-selection plate
to a selection medium supplemented with antibiotics. Incubate for more than one
week.ü@ü@
7. (Day 57; 5
weeks after TF) Select each survived colony on the selection medium as a stable
transformant.
The First
day
[Materials]
üE 200 ml
flask x 1 (autoclave)
üE 300 ml
flask x 2 (autoclave)
üE 10 ml
pipette x 2 (autoclave)
üE 50 ml
centrifuge tube x 2 (autoclave)
üE Funnel
with a sheet of 50 µm nylon mesh (autoclave)
üE Tweezers x
1 (autoclave)
üE Pretreated
cellophane (autoclave: see 2.2)
üE Yellow
tips for P200 pipetteman and blue tips for P1000 pipetteman (autoclave)
üE 0.22
µm syringe-driven filter unit and 10 ml syringe x 2
üE 0.45
µm syringe-driven filter unit and 50 ml syringe x 1
üE Neubauer
hemacytometer (Becton Dickinson no. 424011)
üE Water bath
x 2
üE Centrifuge
üE 50 ml round-bottom
tube (Falcon, Iwaki) (sterile)
üE 15 ml round-bottom
tube (Falcon, Iwaki) (sterile)
üE 15 ml
conical tube (Falcon 2057) (sterile)
üE 6-cm Petri
dish (sterile)
üE Parafilm
[Solution]
üE 500 ml 8%
(w/v) mannitol solution (autoclave)
üE 2 g
PEG6000 in a 10 ml vial and a small stir bar (autoclave)
üE 1% (w/v) MES
(pH5.6)
üE
üE 100 ml protoplast
liquid medium (autoclave)ü@ü@ü@
|
H2O |
90 ml |
|
Stock A* |
1 ml |
|
Stock B* |
1 ml |
|
Stock C* |
0.1 ml |
|
5 g/l Ammonium tartrate |
1 ml (final concentration 50 mg/l) |
|
Mannitol |
6.6 g (final concentration 6.6%) |
|
Glucose |
0.5 g |
|
|
Fill up to 100 ml with H2O |
Stock medium
500
ml 8% (w/v) Mannitol
1
M Ca(NO3)2 Solution
1
M MgCl2 Solution
Driselase
(available from Sigma)
All
of the following stock media are stored at 4˚C.
Stock
A (x 100)ü@ DO
NOT autoclave
|
Ca(NO3)2 4H2O |
118 gü@ ü@(0.5 M) |
|
FeSO4 7H2O |
1.25 gü@ü@ (4.5 mM) |
|
|
Fill up to 1000 ml with H2O |
Stock
B (x 100)ü@ Autoclave
|
MgSO4 7H2O |
25 gü@ü@ (0.1 mM) |
|
|
Fill up to 1000 ml with H2O |
Stock
C (x 100)ü@ü@ Autoclave
|
KH2PO4 |
25 gü@ü@ (1.84 mM) |
|
|
Adjust to pH6.5 with 4M KOH |
|
|
Fill up to 1000 ml with H2O |
[Procedures]
1. Add 0.5 g
of Driselase in a 50 ml conical tube and add 25 ml of 8% mannitol solution. Mix
well.
2. Add 1 ml
of 1 M Ca(NO3)2 and 100 µl
of 1 M Tris-HCl (pH8.0) into 9 ml of 8% (w/v) mannitol solution and mix. Filter
the solution with a 0.22 µm filter.
3. (Preparation
of PEG/T) Add 5 ml of the filtered solution to the autoclaved PEG (see above).
Dissolve PEG completely. This solution is called PEG/T.
4. (Preparation
of MMM) Mix 910 mg of mannitol, 0.15 ml of MgCl2, 1 ml of 1% MES
(pH5.6) and 8.85 ml of H2O. Filter the solution with a 0.22 µm
filter. This solution is called MMM.
5. Switch-on two
water baths at 45˚C and at 20˚C.
6. Centrifuge
the driselase solution at 4,000 rpm for 5 min.
7. Transfer
the supernatant to a 50 ml syringe with a 0.45-µm filter-unit and filtrate
into a 50 ml centrifuge tube.
8. Transfer
the propagated chloronema-rich protonemata (1 to 2 g fresh weight, which are
collected from 10 plates) into the Driselase solution with sterile tweezers. Since
protonemata growing close to the edge of a Petri dish may be contaminated with
bacteria, we collect protonemata more than 1 cm inside from the edge. Then
incubate at 25˚C for 30 min. Mix gently every 5
min.
ü@
9. Filtrate the
protonemata through 50-µm nylon-mesh.

10. ü@Centrifuge the filtrated protoplasts at 1,000
rpm (180 x g) for 2 min at room temperature (22üŗC), and resuspend gently in 40
ml of 8% (w/v) mannitol. Repeat this washing procedure twice.
 ü@
ü@ ü@
ü@
11. ü@Count the number of resuspended protoplasts
with hemacytometer and re-suspend at 1.6 x 106 ml-1 in
MMM solution.
MMM
(ml) = the number of protoplasts per square (large nine squares) x 104
cell/ml x 40 ml / (1.6 x 106 cell/ml)
 ü@
ü@
12. ü@Add 30 µl of plasmid DNA solution into
a 15 ml round-bottom tube (Falcon 2057). Then, add 300 µl of the protoplast
suspension and 300 µl of PEG/T solution
to the tube. Mix gently.
ü@
13. ü@Incubate the tubes containing the
transformation mixture in the 45˚C water bath for 5 min, then in the 20˚C
water bath for 10 min.

14. ü@Dilute the transformation mixture with adding
5 aliquots of 300 µl protoplast liquid medium at
3 min intervals and then 5 aliquots of 1 ml of protoplast liquid medium at 3
min intervals.
ü@ ü@
ü@ ü@
ü@
15. ü@Pour the diluted protoplast solution into a 6
cm Petri dish, seal it with Parafilm, and incubate at 25˚C overnight in
darkness.
ü@ ü@
ü@
[Key points]
üE Resuspend the
protoplasts in the MMM solution with gently pipetting if protoplasts aggregate at
the bottom of the centrifuge tube.ü@
üE Use a round-bottom tube (for example, 14 ml, 17 mm x 100 mm, BD
Falcon tube, no. 352057, Becton Dickinson, Franklin Lakes, NJ) to mix the solution more gently
The second
day
[Materials]
üE 10 ml
disposable pipette (sterile) or white tips for P5000 pipetteman (autoclave)
üE Tweezers x
1 (autoclave)
üE Cellophane (autoclave) – Quality of cellophane is different
depending on companies. For regular cellophane, it is better to be pre-treated
as follows. If you use P-5 cellophane (Futamura Chemical Co., Ltd, Nagoya), we
can use without EDTA treatment. However, other cellophanes may be better to be
treated as follows:
1) Cut
cellophanes to a little bit smaller than the size of a 9 cm-petri dish.
2) Place
cellophane in a 500 ml beaker (usually ** seats) and then add 5 mM EDTA
solution (pH8.0). Autoclave.
3) Wash
with MilliQ water several times. Add MilliQ water in the beaker. Autoclave.
4) Place
cellophane in a glass petri dish and add MilliQ water, then autoclave.
When we use P-5 Cellophane
1)
Cut cellophanes to a little bit smaller than the size of a 9 cm-petri dish.
2)
Place cellophane in a 500 ml beaker and then autoclave.
3)
Place cellophane in a glass petri dish and add MilliQ water, then autoclave
üE 15 ml
conical tube (Falcon)
üE Centrifuge
üE Surgical
tape
[Solution]
üE PRM/T (200
ml); Autoclave and store at 45üŗC
|
H2O |
180 ml |
|
Stock B* |
2 ml |
|
Stock C* |
2 ml |
|
Stock D* |
2 ml |
|
Alternative TES |
0.2 ml |
|
500mM Ammonium tartrate |
2 ml (final concentration 5 mM) |
|
Mannitol |
16 g (final concentration 8%) |
|
CaCl2 2H2O |
0.29 g (final concentration 10 mM) |
|
Agar (Sigma: A6924)* |
1.6 g (final concentration 0.8%) |
|
|
Fill up to 200 ml with H2O |
*
Agar provided by NAKARAI is not appropriate.
üE PRM/B (1,000
ml); Autoclave and pour to 9 cm petri dishes. Dishes are preserved at room
temperature.
|
H2O |
900 ml |
|
Stock B* |
10 ml |
|
Stock C* |
10 ml |
|
Stock D* |
10 ml |
|
Alternative TES |
1 ml |
|
500 mM Ammonium tartrate |
10 ml (final concentration 5 mM) |
|
Mannitol |
60 g (final concentration 6%) |
|
CaCl2 2H2O |
1.47 g (final concentration 10 mM) |
|
Agar (Sigma, A6924, Nacalai Tesque: cat. no. 01028-85) |
8 g (final concentration 0.8%) |
|
|
Fill up to 1000 ml with H2O |
*
Stock medium
All
stock media are stored at 4˚C. Stock D should be used within 2-3 months,
before iron precipitates.
Stock
B (x 100)ü@ Autoclave
|
MgSO4 7H2O |
25 gü@ü@ (0.1 mM) |
|
|
Fill up to 1000 ml with H2O |
Stock
C (x 100)ü@ü@ Autoclave
|
KH2PO4 |
25 gü@ü@ (1.84 mM) |
|
|
Adjust to pH6.5 with 4M KOH |
|
|
Fill up to 1000 ml with H2O |
Stock
D (x 100)ü@ DO
NOT autoclave
|
KNO3 |
|
|
FeSO4 7H2O |
1.25 gü@ü@ (4.5 mM) |
|
|
Fill up to 1000 ml with H2O |
[Procedures]
1. ü@Overlay a sheet of pretreated cellophane on a
9 cm-dish containing PRM/B medium.
2. ü@Transfer the protoplast
suspension into a 15 ml polypropylene conical tube (15 ml, 17 mm x 120 mm, BD
Falcon tube, no. 352196, Becton Dickinson, Franklin Lakes, NJ) with a pipette
and centrifuge at 1,000 rpm (180 x g) for 2min at room temperature
(22üŗC).
3. ü@Discard the supernatant and add 8 ml of a PRM/T
medium stored at 45üŗC. Re-suspend the protoplasts by pipetting.
4. ü@Pour 2 ml of the protoplast suspension on the
pretreated cellophane layered on a PRM/B medium.

5. Seal the
Petri dish using a surgical tape. Incubate the plate at 25˚C for 3 days
under continuous white light with a light flux of 30 - 80 µmol m-2
s-1. We usually use 50 µmol m-2
s-1.
The third
to fifth days
Incubate the plates.
The sixth
day
[Materials]
üE Tweezers x
2 (autoclave)
üE Surgical
tape
[Solution]
üE Selection
medium (BCDAT supplemented with adequate antibiotics)
|
H2O |
900 ml |
|
Stock B |
10 ml |
|
Stock C |
10 ml |
|
Stock D |
10 ml |
|
Alternative TES |
1 ml |
|
500mM Ammonium tartrate |
10ml (final concentration 5 mM) |
|
50mM CaCl2 2H2O (if powder) |
20 ml (final concentration 1 mM) ü@(0.15 g) |
|
Agar (Sigma; A6924, Nacalai Tesque: cat. no. 01028-85) |
8 g (final concentration 0.8%) |
|
|
Fill up to 100 ml with H2O |
After
autoclaving, allow to cool to ~50˚C and add appropriate antibiotics into
the medium.ü@ Store at 4˚C.
Antibiotics
used for selection
a)
Geneticin
(G418)
50
mg/mL solution is available from Invitrogen (no. 10131-035).
Use
at the final concentration of 20 mg/l.
b)
Hygromycin
B
50
mg/mL solution is available from Invitrogen (no. 10687-010).
Use
at the final concentration of 20 mg/l in the medium.
c)
Zeocin
100
mg/mL solution is available from Invitrogen (no. R250-01).
Use
at the final concentration of 50 mg/l in the medium.
!NOTE!
Zeocin is light sensitive. Store zeocin solution and plates or medium containing zeocin in the dark.
d) Blasticidin S
One g of Blasticidin S, Hydrochloride (Funakoshi Co. Ltd KK-400) is dissolved in sterile water in a laminar cabinet, dissolved at 45°C for 10 min, pour 1 mL each into sterile 1.5 ml tubes. This stock solution is preserved at 4°C and use within 6 months. The stock solution is dissolved at 45°C for 10 min and used at the final concentration of 75 mg/L.
e) Nourseothricin/LEXSY NTC
LEXSY NTC (Jena Bioscience, no. AB-101S) is the trade name for the natural product nourseothricin (a complex of the streptothricins F and D) produced by Streptomyces noursei .
Use at the final concentration of 75 mg/l in the medium.
1. ü@Insert tweezers under the cellophane and transfer
the protoplasts all together by lifting the cellophane disc with tweezers to a BCDAT
medium supplemented with adequate antibiotics.
2. ü@Seal the Petri dish with a surgical tape. Incubate
it at 25˚C for 3~4 weeks under continuous white light.ü@
 ü@A
plate after 3-week selection
ü@A
plate after 3-week selection
[Key points]
üE Zeocin is
light sensitive and zeocin plates have to be preserved in the dark before use.
Transfer
to drug-free medium (after an incubation of 3-4 weeks on selection medium)
[Materials]
üE Tweezers
(autoclave)
üE BCDAT
plate
üE Surgical
tape
[Procedure]
1. ü@Transfer each colony under antibiotic selection
to antibiotics-free BCDAT medium. Tweezers are sterilized with 70% alcohol and
fired each time after transfer.ü@
2. ü@Culture at 25˚C for 1 week under
continuous light.
 ü@One-week
colonies on antibiotics-free BCDAT medium after inoculation
ü@One-week
colonies on antibiotics-free BCDAT medium after inoculation
[Key points]
üE Transfer
each colony one by one to a selection medium to avoid generating a chimeric
colony.
üE Incubate
the plate for more than one week.
Transfer
to selection medium (after an incubation of 1 week on drug-free medium)
[Materials]
üE Tweezers (autoclave)
üE BCDAT
plate containing adequate antibiotics
üE Surgical
tape
[Procedure]
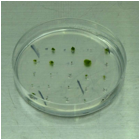
1. ü@After the incubation for one week without
antibiotics, transfer a small part of each protonemal colony to the BCDAT
medium supplemented with adequate antibiotics. A small part of a colony should
be transferred to remove chimeric colonies.
2. ü@Seal the plates with a surgical tape and
incubate for one week.
3. ü@Stable transformants grow on the plate.
 ü@
ü@
[Key points]
üE Transferred
protonemata should be collected from the leading edge of a colony to prevent
collecting chimeric clones. Do not transfer a whole colony or gametophores.
üE When using
plates containing zeocin, ensure they are freshly prepared.
üE Some kinds
of disruptants may show reduced growth. Do not miss positive transformants even
if growth of the colony is reduced.
üE PEG-mediated transformation generates polyploids by protoplast fusion. Morphology of polyploid protonemata is not distinguished from that of wild type. DNA amount of transformants should be examined using flow cytometry (see chapter 8. Flow Cytometry Analysis).