
|
|
LABORATORY OF GENOME INFORMATICS |
| Research Associate: | UCHIYAMA, Ikuo |
|
Accumulation of biological data has recently been accelerated by various high-throughput so-called “ omics“ technologies such as genomics, transcriptomics, proteomics and so on. The field of genome informatics is aimed at utilizing these data, or finding some principles behind the data, for understanding complex living systems by integrating the data with current biological knowledge using various computational techniques. In this laboratory we focus on developing computational methods or tools for comparative genome analysis, which is a useful approach for finding functional or evolutionary clues to interpreting genomic information of various species. Especially, the current focus of our research topics is on comparative analysis of microbial genomes, the number of which is now beyond a hundred, as a basic model system for understanding the variety of life through the comparative analysis of numerous genomic sequences simultaneously. I. Construction of microbial genome database for comparative analysisThe number of completed microbial genome sequences is growing rapidly, and nearly two hundreds genome sequences in various levels of relatedness have already been available today. The role of comparative genomics becomes much more important to utilize these large number of sequences not only for elucidating commonality in all of life, but also for understanding the evolutionary diversity within various groups, as well as for understanding the evolutionary processes or mechanisms producing such diversity. We have been developing and maintaining a database system for comparative analysis of microbial genomes named MBGD (http://mbgd.genome.ad.jp/). The central function of MBGD is to create orthologous groups among multiple genomes (Figure 1), which is a crucial step for comparative genome analysis.The key components of MBGD include i) an algorithm that can classify genes into orthologous groups using precomputed all-against-all similarity search results, ii) a user interface that is designed for users to explore the resulting classification in detail, and iii) an incremental updating process for similarities between genes and other data, which enables the system to provide the latest data rapidly. By this approach, MBGD is now the world’s largest database of its kind. Moreover, by specifying a set of organisms, users can obtain appropriate classification results that they want using the latest data available. This unique feature is especially useful for users whose interests are focused on some taxonomically related organisms. Indeed, we used this feature of MBGD in our comparative genomics studies among Bacillus-related species described below (see Figure 1). |
A 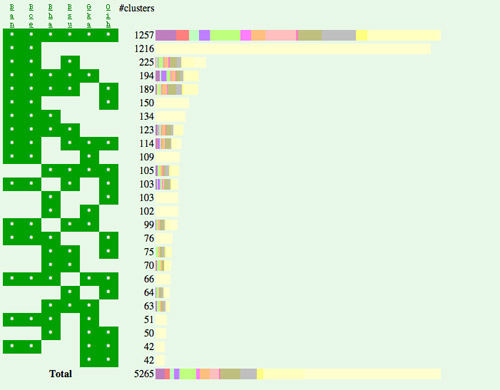 |
B 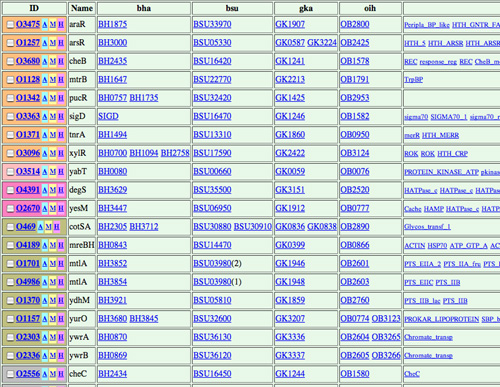 |
Figure 1 Ortholog cluster map (A) and ortholog cluster table (B) in MBGD. Here, the orthologous grouping was created using genomes of 6 Bacillus-related species including G. kaustophilus. In the ortholog cluster map (A), the grouping result is sorted according to the phylogenetic patterns, which represent presence or absence of the orthologs in each genome, and each cluster is assigned a color according to its function category. By clicking the bar graph of the map, the actual cluster table is shown (B), where each row represents an ortholog cluster and each column represents an organism. |
|
We are also involved in a reannotation project for bacterial genomes that is currently in progress by the Japanese research community of pathogenic bacterial genomics (headed by Dr. Kuhara, Kyushu University). In this project, an orthologous grouping created by the MBGD system is used as a prototype grouping, which will be further modified and reannotated manually. |
II. Revealing thermoadaptation trait by comparative genomics between thermophilic Geobacillus kaustophilus and mesophilic Bacillus related species.How thermophilic organisms adapt to high-temperature environments has long been an intriguing question in both academic and industrial fields. Recent studies revealed some remarkable genomic features strongly correlated with thermophily such as amino acid composition, codon usage and genomic dinucleotide composition. However thermophiles whose genomic sequences have already been determined are somewhat phylogenetically biased; many of them belong to archaea or the deep-branching bacteria (Aquifex and Thermotoga). On the other hand, one of the effective approaches in revealing thermophilic trait is to compare genomes between closely related organisms including both thermophiles and mesophiles. This approach is also effective for understanding thermoadaptation from the viewpoint of evolution, although the genomic sequences from an appropriate set of organisms are needed, which have not yet been obtained. In collaboration with Dr. Takami’s group (JAMSTEC), we determined the complete genomic sequence of thermophilic Geobacillus kausophilus and compared it with those of phylogenetically related 5 mesophilic bacilli, Bacillus subtilis, Bacillus halodurans, Oceanobacillus iheynsis, Bacillus cereus and Bacillus anthracis.By the principal component analysis of the amino acid composition using 150 prokaryotic genomes including 20 thermophilic ones, we were able to find that the thermophilic G. kaustophilus were distinguishable from mesophilic bacilli by the borderline distinguishing thermophiles from mesophiles on the second principal axis, although on the whole the Bacillus-related species were located near this borderline (Figure 2). Further statistical analysis revealed some asymmetric amino acid substitutions between the thermophilic and the mesophilic bacilli, which are possibly associated with the thermoadaptation of the organism. In addition, upon orthologous grouping of the 6 bacillar sequenced genomes, we found 839 genes (24%) in the G. kaustophilus genome being unique to that species, among which we were able to find some candidate genes that may contribute to thermophily presumably by enhancing the stability of nucleic acids. |
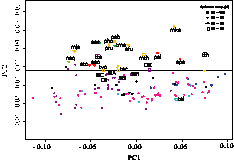 |
Figure 2 Principal component analysis of the amino acid composition using 150 prokaryotic genomes. Thermophiles are marked by symbols according to their optimal growth temperatures. An abbreviated name in upper case is assigned to each Bacillus-related species; GK: G. kaustophilius, BH: B. halodurans, BS: B. subtilis, BA: B. anthracis, BC: B. cereus, OI, O. iheyensis. An abbreviated name in lower case is assigned to each thermophile. |
III. Identification of the common core structure of bacillar genomesRecently several studies showed that horizontal transfer as well as vertical transfer has played important roles in the prokaryotic genome evolution. However, to obtain clearer picture of the bacterial genome evolution, we need further detailed investigations including extensive comparison of multiple genomes that are closely or moderately related to each other. In the collaborative work with Dr. Takami, we also performed such extensive comparison among Bacillus-related genomes for the purpose of drawing general picture of the bacterial genome evolution. Aerobic endospore-forming gram-positive Bacillus-related species is known to be able to grow in a wide range of environments. The above-mentioned 6 bacilli whose genomic sequences have been determined include alkaliphilic B. halodurans, halotolerant O. iheyensis and thermophilic G. kaustophilus, in addition to well-known laboratory strain B. subtilis and pathogenic B. anthracis and B. cereus. These organisms except B. anthracis and B. cereus are moderately diverged each other and are belonging to distinct major clusters in the 16S rRNA phylogenetic tree. By simple pairwise dotplot analyses between them, one can easily see some large collinear regions along the diagonal lines of the plots so that the overall genomic structures are primarily well conserved between organisms. However, one can also find a substantial number of species-specific genes that are inserted in each of the genomes. To investigate further, we are trying to identify common “core structure“ of bacillar genomes, which is defined as a set of sufficiently long consecutive genomic segments in which gene orders are conserved among multiple genomes so that they are likely to have been inherited from the common ancestor mainly through vertical transfer. We have developed a novel algorithm for aligning conserved regions of multiple genomes by sorting orthologous groups so as to retain the conserved gene orders as many as possible. From this alignment, we were able to identify the common core structure of bacillar genomes comprising about 1500 genes. It appears that most of the important genes are included in the resulting core gene set. For example, the set contains 246 out of 271 B. subtilis essential genes that were primarily determined by a systematic inactivation experiment. Further characterization of the core gene set is currently in progress. |
Publication List:Takami, H., Takaki, Y., Chee, G-J., Nishi, S., Shimamura, S., Suzuki, H., Matsui, S., Uchiyama, I. (2004) Thermoadaptation trait revealed by the genome sequence of thermophilic Geobacillus kaustophilus. Nucleic Acids Res., 32, 6292-6303. |
 |